Non-B DNA structures and their contributions to genetic diversity, aging, and disease
Eleftherios Bochalis, Irene Dereki, Guliang Wang, Argyro Sgourou, Karen M Vasquez, Ilias Georgakopoulos-Soares

TL;DR
This paper explores how alternative DNA structures contribute to genetic diversity, disease, and aging by affecting genome stability and function.
Contribution
The paper provides a comprehensive overview of recent advances in understanding the biological roles and consequences of non-B DNA structures.
Findings
Non-B DNA structures are enriched in functional genomic regions and are linked to gene regulation and genome instability.
These structures contribute to replication stress, transcription stalling, and DNA breaks, leading to mutational hotspots.
Non-B DNA structures play a dual role in promoting genetic variation and contributing to mutations in aging and disease.
Abstract
DNA is most often found in its canonical B-form double-helical structure, but can also adopt alternative conformations, known as non-B DNA structures. Numerous non-B structures have been characterized, including G-quadruplexes, i-motifs, Z-DNA, hairpins, cruciforms, slipped structures, R-loops, and H-DNA. Non-B DNA motifs are enriched in functional regions, including near transcription start and end sites, topologically associated domains, and replication origins, suggesting their importance in gene regulation, genome organization, and replication. However, these structures are intrinsically prone to error-generating processing, leading to genomic instability and hence have been implicated in the development of human diseases. Here, we discuss recent advances in understanding the biological roles of non-B DNA structures and their contribution to genomic instability in somatic and…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
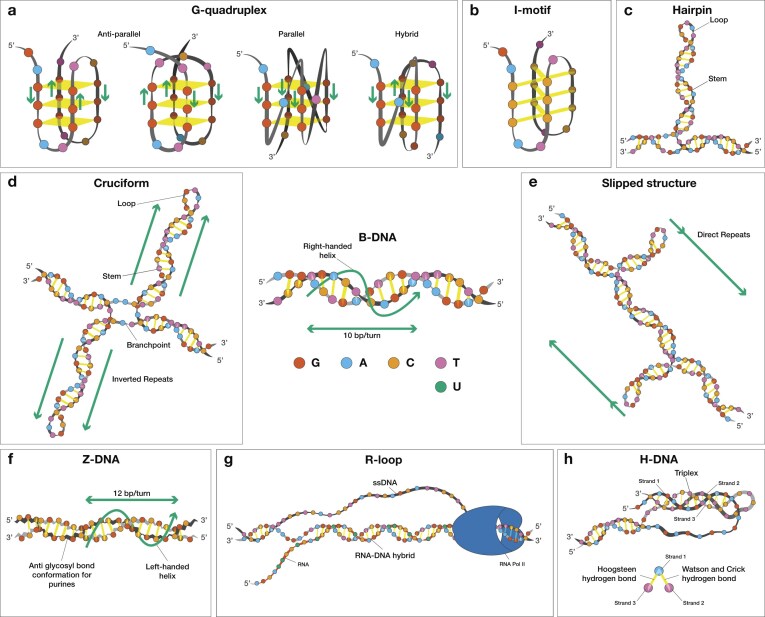 Figure 1
Figure 1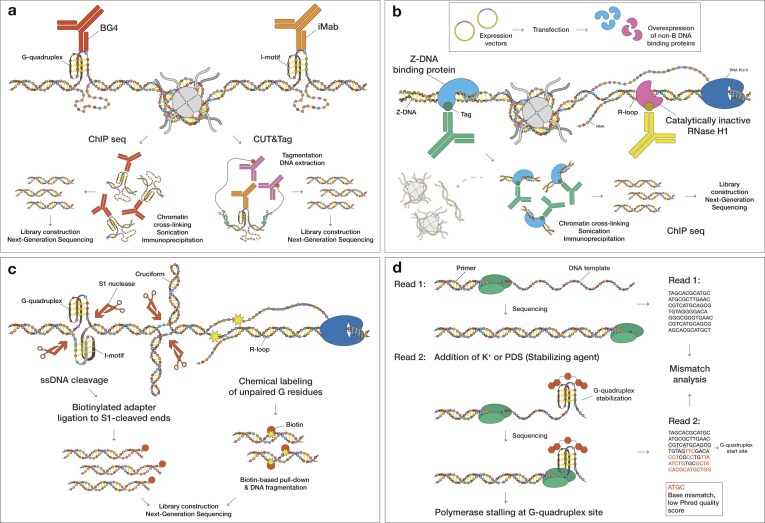 Figure 2
Figure 2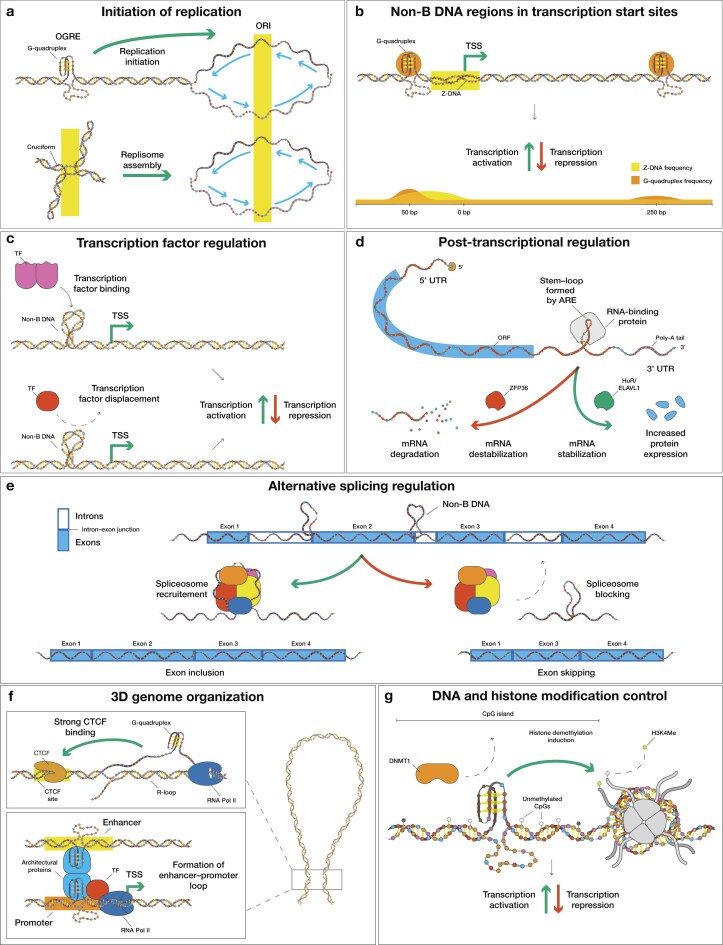 Figure 3
Figure 3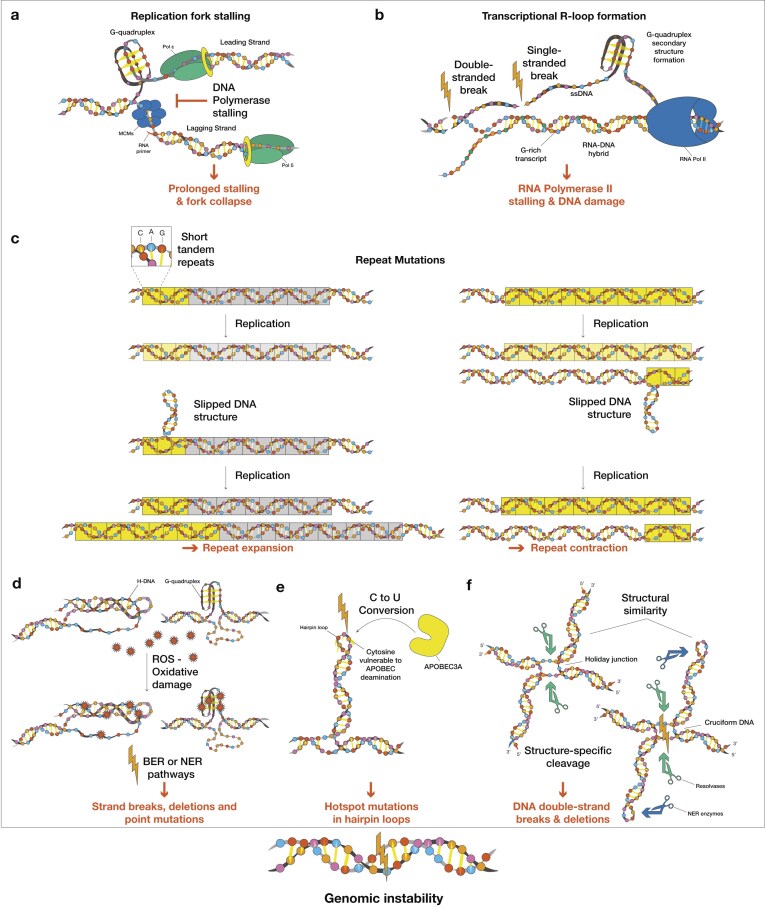 Figure 4
Figure 4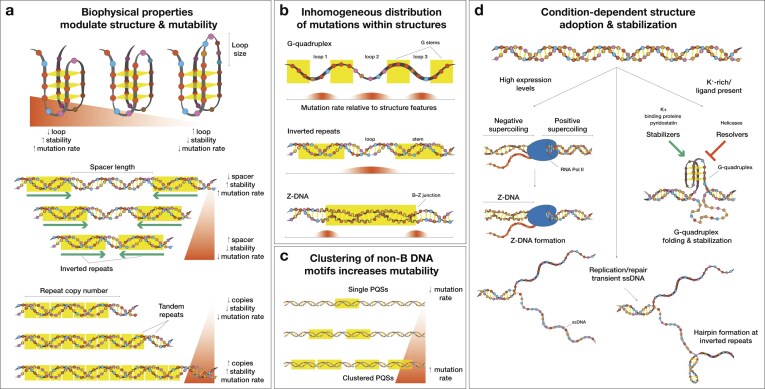 Figure 5
Figure 5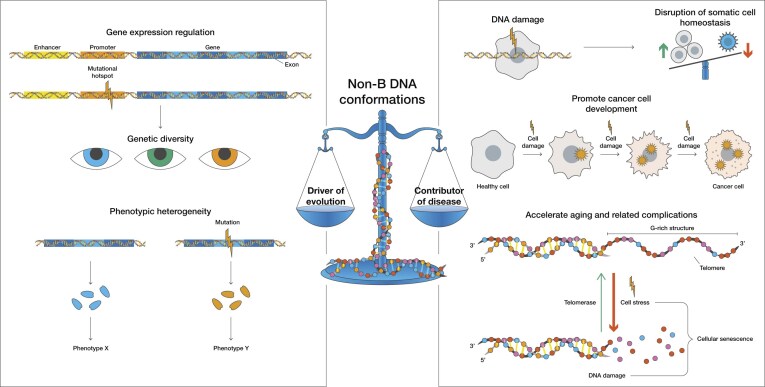 Figure 6
Figure 6- —National Institute of General Medical Sciences10.13039/100000057
- —National Institutes of Health10.13039/100000002
- —National Cancer Institute10.13039/100000054
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsDNA and Nucleic Acid Chemistry · DNA Repair Mechanisms · Advanced biosensing and bioanalysis techniques
Introduction
The canonical conformation of the DNA molecule, initially described by Watson, Crick, Wilkins, and Franklin in 1953 [1], is a right-handed double helix with a diameter of ∼2 nm (Fig. 1). Under typical cellular conditions, this is the predominant and most thermodynamically stable structure, commonly termed B-DNA. However, specific sequence motifs, ionic environments, and topological stresses can result in the formation of noncanonical or alternative structures (i.e., non-B DNA) [2–4]. Non-B DNA structures have been implicated in gene regulation, genome organization, recombination, replication, and responses to environmental stresses, among other biological processes [5–11]. Loci that are predisposed to non-B DNA formation include repetitive sequences capable of adopting G-quadruplex DNA, Z-DNA, hairpin/cruciform inverted repeats, slipped DNA structures formed at direct and tandem repeats, and H-DNA mirror repeats (Fig. 1). Non-B DNA structures require negative supercoiling to form; hence, processes such as replication, transcription, and DNA repair promote their formation [12–14]. These motifs are enriched in open chromatin regions, promoters, 5′ and 3′ untranslated regions, near transcription start and end sites, among other regions, and are often associated with transcriptionally active loci [5, 9, 11, 15, 16].
Schematic of non-B DNA structures. Schematic illustrations of noncanonical DNA conformations, beyond the physiological B-DNA form. (A) G-quadruplex structure in which stacked guanine tetrads are held together with Hoogsteen hydrogen bonds. (B) I-motif structure. (C) Hairpin, and (D) cruciform formation at inverted repeats. (E) Slipped structure formation at tandem repeats. (F) Z-DNA structure. (G) R-loop formation during transcription. (H) H-DNA formation at mirror repeats stabilized by Hoogsteen hydrogen bonds. A Hoogsteen base pair is a type of non-Watson–Crick bond, where the purine base rotates 180° with respect to the helix axis and adopts a syn conformation. B-DNA is depicted at the center of the figure, showing the right-handed structure with 10 bps per turn.
Despite their important regulatory functions, these same non-B DNA structures present a double-edged sword for genomic stability. Mutations in the human genome are not distributed homogeneously [17–20]. Non-B DNA-forming sequences can serve as endogenous mutational hotspots [19, 21], due to their intrinsic genomic instability, and can lead to functional consequences. Such non-B DNA-induced instability has been shown across prokaryotic and eukaryotic organisms [22–27] and has been associated with disease development, including cancer and neurodegenerative disorders in humans [19, 27–34].
Beyond their role in disease, recent evidence suggests that non-B DNA-induced mutagenesis can also affect the process of aging. In somatic cells, in vivo models show that non-B DNA structures act as a source of local instability that can increase with age in a tissue-specific manner [35–37]. Such age-associated increases in mutational burden and/or apoptosis can disrupt tissue homeostasis and drive cellular dysfunction, ultimately contributing to the aging process [38, 39]. In germline cells, the same propensity for genetic instability can serve as a template for genetic variation, thereby facilitating evolutionary adaptation. This dual nature underscores the importance of elucidating the functional roles of non-B DNA, both to advance our understanding of the mechanisms of aging and complex diseases, and to enable more precise modeling of evolution [8, 40, 41].
In this perspective, we examine recent advances in understanding the roles of non-B DNA motifs in both genome function and dysfunction. We highlight how recent developments in sequencing technologies can facilitate the examination of non-B-DNA-forming sequences in low-complexity and highly repetitive genomic regions. These previously challenging loci open new avenues for understanding the roles of non-B DNA in disease and evolution. Finally, we examine recent results on the association of non-B DNA with somatic mutations in aging, cancer, and neurodegenerative disorders, and propose directions for leveraging non-B DNA research in therapeutic and diagnostic applications.
Non-B DNA categories and their detection
Specific sequence features, including guanine-rich tracks, alternating purine-pyrimidine runs, mirror, inverted, and tandem repeats, predispose DNA to adopt non-B conformations [6]. G-quadruplexes are nucleic acid structures stabilized by Hoogsteen hydrogen bonds between guanines that form stacked G-tetrads [42, 43] (Fig. 1A). Their cytosine-rich complementary sequence can fold into intercalated motifs (i-motifs) (Fig. 1B). However, G-quadruplexes and i-motifs may not coexist due to steric hindrance; the possibility of their coexistence increases with the offset distance [44]. DNA hairpins can form at both perfect and imperfect inverted repeats. These repeats consist of two adjacent sequences, one of which is the reverse complement of the other. In hairpin structures, the complementary arms are held together by hydrogen bonds, while the spacer region remains single-stranded (Fig. 1C). A related structure is the cruciform, which resembles the Holliday junction that forms during recombination and is composed of two hairpins and a four-way junction [45, 46] (Fig. 1D). Slipped structures form at consecutive repeat sequences, in which one repeat unit misaligns with the second repeat unit on the opposite strand, therefore creating single-stranded DNA (ssDNA) and exposed loops [47] (Fig. 1E). Z-DNA is a left-handed double-helical structure typically formed at alternating purine and pyrimidine sequences [48, 49] that contains ∼12 bps per helical turn instead of the canonical 10 bps in B-DNA. The point at which a B-DNA motif transitions to a Z-DNA conformation, and vice versa, is termed a B-Z junction, which contains unpaired bases. (Fig. 1F). R-loops are three-stranded nucleic acid structures formed when an RNA molecule hybridizes with one strand of DNA, leaving the complementary DNA strand unpaired [50]. They can occur naturally during transcription and play roles in gene regulation; however, excessive R-loop formation can lead to genomic instability and has been associated with several human diseases [51, 52] (Fig. 1G). Another non-B DNA structure is intramolecular triple-stranded DNA (H-DNA), which can form at homopurine-homopyrimidine sequences that exhibit mirror repeat symmetry [53]. In this structure, one DNA strand folds back into the major groove of the underlying double helix and is stabilized by Hoogsteen or reverse-Hoogsteen hydrogen bonds, forming a triple-helical DNA structure with the complementary strand left unpaired (Fig. 1H). Intermolecular triplexes can also form between DNA molecules, RNA molecules, or between a DNA–RNA hybrid [54]. In each case, Hoogsteen or reverse-Hoogsteen hydrogen bonds stabilize the triplex structure. When the third strand is an independent oligonucleotide, it is termed a triplex-forming oligonucleotide.
Various in vitro and in vivo methods have been developed for the detection of non-B DNA-forming sequences [55, 56]. Some techniques use antibodies [57–59] (e.g., BG4 for G-quadruplexes) [60, 61]; iMab for i-motifs [62, 63] (Fig. 2A), binding proteins (e.g., Z-DNA binding domain of ZBP1 or ADAP1) [64, 65], or inactivated enzymes (i.e., “dead” RNase H for R-loop detection) [66, 67] (Fig. 2B). Also, structure-agnostic techniques exist, which rely on the ssDNA or distorted DNA regions for non-B DNA detection [11, 68, 69] (e.g., S1-END-seq), while others can label ssDNA exposed during transient structural transitions [70] (e.g., KAS-seq) (Fig. 2C). Assays that capture transient ssDNA can provide a high-resolution, genome-wide map of regions prone to adopting non-B DNA conformations [71]. In G4-seq [72, 73], treatment with G-quadruplex-stabilizing or -destabilizing ions/small molecules has been shown to affect the in vitro folding potential (Fig. 2D). However, these methods have limitations. Antibody- and ligand-based approaches exhibit binding biases and can sometimes stabilize or destabilize non-B DNA structures [54, 60, 74–76]. Certain antibodies are biased toward sequence recognition and secondary structure identification [77–79]. Run et al. (2025) recently summarized different methods used for detecting Z-DNA and discussed their strengths and limitations [80].
Non-B DNA detection methods. (A) Non-B DNA structure-specific antibodies (e.g., BG4 and iMab) enable genome-wide mapping either by immunoprecipitation of antibody-bound, crosslinked chromatin region (ChIP-seq) or by antibody-guided tagmentation at bound sites (CUT&Tag). (B) Catalytically inactive or binding-only protein probes with specificity for non-B DNA (e.g., the Z-DNA-binding domain of ZBP1 and catalytically dead RNase H1) linked with an epitope tag are expressed in cells. Non-B DNA motifs are mapped by anti-tag ChIP-seq. (C) ssDNA footprinting approaches: in S1-END-seq, the ssDNA-specific nuclease S1 cleaves non-B DNA-associated ssDNA to generate double-strand breaks that are adapter-ligated and sequenced; in KAS-seq, ssDNA is labeled with N3-kethoxal, followed by click-mediated biotinylation, streptavidin pull-down, and sequencing to map R-loop-associated ssDNA. (D) In G4-seq, G-quadruplex-stabilizing molecules are used to ensure G-quadruplex formation. Such formation can result in polymerase stalling, thereby reducing sequencing quality.
Direct detection of non-B DNA structures in the genomes of living cells is even more challenging because they are dynamic and transient. Nevertheless, certain newer approaches have been developed to study this dynamic nature. For R-loops, a genetically encoded sensor consisting of three RNase H1 domains linked to a fluorescent protein can be used for live-cell imaging of cellular R-loops and for the kinetic analysis of R-loop formation [81]. For G-quadruplexes, Galli et al. (2022) have developed a fluorophore-linked nanobody that can be expressed intracellularly and detect G-quadruplexes in living cells [82]; Di Antonio et al. (2020) have engineered a fluorescent probe that, at low concentrations, enables single-molecule and real-time detection of individual G-quadruplex structures [83]; and Summers et al. (2021) have developed a fluorescence lifetime imaging microscopy assay to study G-quadruplex formation in real time [84]. Finally, in-cell nuclear magnetic resonance (NMR) has been employed to study the equilibrium between i-motifs and B-DNA during replication in living cells [85]. Again, most of the techniques mentioned above provide either indirect measurements or require intermediate steps (e.g. cell fixation), which can stabilize or destabilize existing non-B DNA structures [6]. Therefore, it is important to consider that the non-B DNA conformations detected by these experimental methods reflect only the specific conditions under which they were tested.
Experimental techniques have enabled the development of computational algorithms to detect G-quadruplexes [86–95], Z-DNA [96–99], R-loops [100, 101], H-DNA [102–104], and i-motifs [105, 106]. They use sequence motifs, thermodynamic parameters, and machine learning techniques to systematically scan genomes for loci whose sequence composition and predicted thermodynamic stability resemble those of experimentally validated sequences. The experimental data informing these predictions consist primarily of in vitro biophysical measurements on purified oligonucleotides and genome-wide non-B DNA maps. These tools can analyze regions inaccessible to experimental methods and enable systematic genome-wide predictions. Beyond stand-alone algorithms, many databases have been created that compile experimentally validated and computationally predicted non-B DNA-forming sequences, offering a valuable resource for integrative analyses across genomes [100, 107–115]. Despite their utility, they have certain drawbacks. Most tools cannot handle cellular contexts (salt concentrations, protein interactions, and chromatin state), whereas different algorithms designed for the same non-B DNA structure can yield inconsistent predictions for the same sequence due to differences in parameters utilized. They typically provide static predictions and cannot capture the transient, dynamic nature of non-B DNA formation; moreover, machine learning approaches may inherit biases from the experimental data used for training.
Note: While these tools can handle high-throughput data at scale and at low cost, it is crucial to keep in mind that they only predict a sequence’s potential to form a non-B DNA structure, and hence experimental validation is required to verify this prediction. When interpreting experimental findings, it is essential to point out that terms such as non-B DNA structures and non-B DNA-forming sequences are often used interchangeably. Careful evaluation is advised to determine whether studies provide evidence for DNA structure formation or for sequences with the potential to form such structures. Evidence for structure formation should also be considered in the context of the experimental design used. Factors such as ionic strength and superhelical tension can vary significantly between in vitro and in cell-based assays. Additionally, non-B DNA structures are highly dynamic; a noncanonical structure may form in one cellular state (e.g., during replication) but be absent in another, depending on factors such as local negative supercoiling and chromatin accessibility.
Non-B DNA at regulatory hotspots drives genome function
Non-B DNA-forming sequences are inhomogeneously distributed across the human genome [19]. While tandem repeats are primarily found in highly repetitive regions of the genome, most non-B DNA motifs are preferentially positioned and enriched at cis-regulatory elements, telomeres, centromeres, and introns.
Near or within origins of replication (ORI), non-B DNA-forming sequences can influence replication in two opposing ways: they can activate DNA replication, yet they can also impede polymerase progression [116–118]. On one hand, an origin G-rich repeated element (OGRE), known to form G-quadruplex structures, facilitates replication initiation in mammalian genomes, while its deletion strongly reduces origin firing [116, 119] (Fig. 3A). On the other hand, i-motif structures in the template strand can cause polymerases to stall more efficiently than G-quadruplexes [120]. G-quadruplex and i-motif structures are mutually exclusive in most cases, despite originating from complementary strand contexts. During replication, their formation is regulated by a cell-cycle-dependent “switch”. G-quadruplexes peak during S phase [57], while i-motifs reach their maximum during late G1 phase [62, 121]. Inverted repeats, and especially cruciform-forming sequences, have been found to act as triggering signals for replication initiation, when placed in ORI sites [122] (Fig. 3A). Non-B DNA motifs can also stall polymerase complexes. For example, a Z-form DNA–RNA hybrid, which can be stabilized by CpG methylation, was found to inhibit the initiation and elongation of Okazaki fragments [123]. In mammalian cells, H-DNA-forming repeats can antagonize ORI activation by limiting helicase-driven duplex unwinding. Finally, the H-DNA-induced stalling is more prominent when the non-B DNA-forming sequence is present on the lagging strand [124].
Consequences of non-B DNA structures present at regulatory elements. (A) G-quadruplexes located at OGREs promote origin firing and replisome assembly. (B) G-quadruplexes and Z-DNA near transcription start sites (TSSs) bias transcriptional initiation and pausing, thereby increasing transcriptional activation. (C) Non-B DNA-forming sequences adjacent to transcription factor (TF) binding sites can either recruit or displace TFs. (D) Hairpins formed at AU-rich elements (AREs) aid RNA-binding protein (RBP) binding and can either stabilize mRNA or accelerate mRNA decay. (E) Non-B DNA conformations near splice junctions alter spliceosome recruitment, driving exon inclusion or skipping. (F) R-loops can co-localize with G-quadruplexes, enhancing CCCTC-binding factor (CTCF) binding and supporting long-range enhancer–promoter looping via YY1 and RNA Pol II. (G) Non-B DNA motifs within CpG islands can limit DNMT1 activity, leading to hypomethylated states and promoting histone demethylation.
Non-B DNA-forming sequences are also involved in the transcriptional process, and often accumulate near or within promoters, enhancers, insulators [11, 21, 62, 125–132], TSSs, and transcription end sites (TESs) [21, 133–135]. Studies have shown that non-B DNA motifs can regulate gene expression through multiple mechanisms, including controlling transcription factor binding, nucleosome positioning, epigenetic modification regulation, and modulating splicing and translation [11, 21, 62, 125–132, 136–139] (Fig. 3B). Non-B DNA structures within or adjacent to transcription factor binding sites can either aid in transcription factor recruitment and stabilization, or displace transcription factors entirely [16, 21, 139] (Fig. 3C). For example, the interaction of YY1 with G-quadruplexes can aid DNA looping [140] and recognition of non-B DNA structures by p53 [141], whereas reporter plasmid assays show G-quadruplexes within the KRAS promoter reduce its transcriptional efficacy by ∼20% [142]. An i-motif-forming sequence present in the mid-region of the KRAS promoter is recognized by the transcription factor hnRNP K, which positively modulates KRAS transcription [143]. In the BCL2 promoter, an i-motif structure exists in equilibrium with a hairpin conformation, where the i-motif structure can be detected by hnRNP LL, leading to transcriptional activation [144, 145]. The presence of i-motif sequences in the MYC promoter was shown to diminish gene expression [146], whereas G-quadruplex formation has shown mixed outcomes. For instance, a recent study demonstrated that CRISPR-dCas9 fused to nucleolin targeting the MYC P1 promoter G–quadruplex-forming sequence effectively induced G-quadruplex folding, resulting in transcriptional repression of MYC [147]. Another study genetically abrogated endogenous G-quadruplex structure formation at the MYC P1 promoter, resulting in reduced expression [148]. G-quadruplexes can also mediate distant enhancer–promoter interactions. Using a CRISPR-Cas9 system, Doyle et al. (2025) demonstrated that G-quadruplex-forming sequences present in the α- and β-globin enhancer loci are necessary to sustain active chromatin, RNA Pol II recruitment, and enhancer–promoter interactions. Consistent with these findings, disruption of the G-quadruplex-forming sequence hinders globin transcription, whereas replacement with an unrelated G-quadruplex-forming sequence restores enhancer function [149]. Z-DNA structures can have a dual effect on transcription regulation. On one hand, Z-DNA-forming sequences have been found to drive increased expression when placed upstream of genes [21]. On the other hand, in vitro results have shown that the RNA polymerase can partially stall at the B-Z junction when moving from a B-DNA segment to a Z-DNA segment [124, 150]. H-DNA formation can also impede transcription. During RNA polymerase elongation, additional negative supercoiling is generated behind the enzyme, promoting non-B DNA formation at H-DNA-forming sequences. This in turn creates a sharply bent DNA structure that can lead to polymerase stalling [151].
AREs are the most common cis-regulatory elements located within 3′ untranslated regions (UTRs), and are capable of hairpin structure formation [152–154]. Such hairpin structures are recognized by RBPs and play a crucial role in regulating messenger RNA (mRNA) decay at the post-transcriptional level. HuR/ELAVL1 and ZFP36 are two examples of RBPs that bind hairpin AREs and function in opposite ways. HuR/ELAVL1 is a mRNA stabilization factor that antagonizes mRNA decay by blocking access of destabilizing factors, limiting recruitment of the decapping machinery, leading to increased protein output [155–158] (Fig. 3D). In contrast, ZFP36 recognizes the same hairpin AREs, but recruits the CCR4-NOT deadenylase to drive mRNA decapping, resulting in faster decay and minimized protein expression [159–163] (Fig. 3D). Transcriptome-wide PAR-CLIP analysis revealed that over 80% of ZFP36 sites in 3′ UTRs overlapped with HuR/ELAVL1 target sites. ZFP36 exhibited higher affinity for AREs compared to HuR/ELAVL1 [164], and in vitro models show that ZFP36 has very low basal expression, with HuR/ELAVL1 being more abundant in cells. Alternative pathways for 3′ UTR hairpin-mediated mRNA decay have also been identified [160]. Finally, non-B DNA-forming sequences are enriched near splice junctions, exhibiting either recruitment or repulsion of splicing factors [165–167] (Fig. 3E).
The inherent genomic instability of non-B DNA can facilitate biological processes such as gene regulation and recombination. For example, co-transcriptionally formed R-loops have been shown to promote recombination [168, 169] and immunoglobulin class switch recombination [170]. Z-DNA-forming sequences serve as markers for the AIRE transcription factor to select genes that drive thymic T cell tolerization, through DNA double-strand break (DSB) formation and promoter poising [136]. G–quadruplexes in immunoglobulin switch regions have been shown to enhance AID targeting, thereby promoting class–switch recombination [171, 172]. Non-B DNA-forming sequences are also enriched at recombination sites, including at binding sites of PRDM9, a protein responsible for specifying the genomic sites of meiotic recombination in mammals [173–176]. H-DNA-forming sequences have also been shown to induce homologous recombination in mammalian cells, with H-DNA-forming oligonucleotides often discussed as potential genome editing tools [177–179]. In addition, cruciform-forming sequences can lead to DNA rearrangements by both replication-dependent and -independent mechanisms [23].
Non-B DNA can also contribute to higher-order genome organization and epigenetic regulation. G-quadruplexes on the G-rich strand can facilitate the formation of R-loops on the complementary strand at CTCF sites, promoting CTCF binding and hence genome architecture organization [130, 180–182] (Fig. 3F). Attenuating R-loop formation has been shown to reduce CTCF occupancy and alter gene expression. G-quadruplex-forming sequences are also enriched at topologically associated domain boundaries [183]. Non-B DNA structures help bridge distal elements. As previously mentioned, the presence of G-quadruplex-forming sequences in enhancers and promoters facilitates identification by YY1, and subsequent YY1 dimerization and mediation of long-range enhancer–promoter looping [184] (Fig. 3F). It has been proposed that enhancers and promoters can form G-quadruplexes cooperatively, with each region contributing “half of a G-quadruplex”, leading to distant DNA looping without the need for architectural proteins [185, 186]. G-quadruplex structures identified within CpG islands have been found to inhibit DNMT1 enzymatic activity, leading to local hypomethylation [187, 188] (Fig. 3G). R-loop formation in promoter CpG islands likewise protects against DNA methylation from DNMT3B [131, 189], and both in vitro and in vivo studies suggest that Z-DNA structures inhibit DNMT3A [190, 191]. Furthermore, promoter G-quadruplex-forming sequences and associated proteins (i.e., NME2) can recruit the REST-LSD1 complex and induce histone H3 Lys4 demethylation [9] (Fig. 3G). Other non-B DNA-forming sequences have been found to affect nucleosome placement. In vitro and in vivo results show that G-quadruplex-forming sequences are located at the deepest point of nucleosome exclusion at promoters and correlate with maximum promoter activity [192]. H-DNA formation is thought to be inhibitory to nucleosome assembly, leading to a bias toward H-DNA exclusion from nucleosomes [193]. Z-DNA-forming sequences can also be refractory to nucleosome assembly [194], and hence they can not be incorporated within histone cores, except at their termini [195]. However, these inhibitory effects are not universal across non-B DNA-forming sequences; some long triplet repeat sequences, that can adopt hairpin structures, have instead been found to be strong nucleosome positioning elements [193].
Because non-B DNA-forming sequences occur in repetitive genomic regions, they are often found in telomeric and centromeric loci. Telomeric TTAGGG tracks primarily adopt T-loop formation. During replication or TERRA transcription, these T-loops are broken and can form G-quadruplexes, i-motifs, or R-loops, which assist in telomere capping and maintenance. However, if these non-B DNA structures are not properly resolved, they can induce DNA damage leading to recombination-based telomere lengthening and end-end fusions [196–199]. Telomere-to-telomere genomic assemblies of six great apes have shown substantial enrichment for non-B DNA-forming sequences in centromeric regions, with inverted repeats displaying moderate enrichment at approximately half of all centromeres analyzed [200, 201]. It is also proposed that centromeres form at non-B DNA motifs and can be stabilized by sequence-specific DNA-binding proteins [40, 202].
Non-B DNA across the tree of life
Non-B DNA-forming sequences are often enriched at functionally important genomic sites, though differences can be found across the tree of life. Analysis of more than 100,000 genomes showed that G-quadruplex-forming sequences are unevenly distributed across the three domains of life and viruses, with lineage-specific preferences. Despite this variability, their genomic placement is nonrandom, with cis-regulatory elements exhibiting higher enrichment. For example, in bacteria belonging to the Deinococcota phylum, G-quadruplexes are preferentially placed upstream and downstream of TSSs and TESs, respectively [108]. Near splice sites, G-quadruplex motifs are predominantly found in certain lineages, such as vertebrates and birds, but are depleted in others [165]. Across great apes, G-quadruplexes are also enriched at enhancers, promoters, UTRs, and ORI, but depleted at protein-coding regions. Also, short tandem repeats are enriched in the repetitive telomeric and centromeric regions [201]. For both motifs, additional species-specific enrichments are observed [203]. In yeast, transcription-replication head-on collisions during meiosis lead to de novo R-loop formation, which may promote meiotic DSBs [204].
Cruciform/hairpin-forming inverted repeats are the most abundant non-B DNA promoter motif in prokaryotes [205, 206], while in the case of Vibrio parahaemolyticus, a pathogenic Vibrio species, cruciform elimination by the HlyU protein enables concomitant virulence gene expression [207]. Inverted repeats can also drive Rho-independent transcription termination and are often found in terminator regions [208]. In bacterial biofilms, G-quadruplexes and Z-DNA protect from nuclease degradation [209]. Across most viral species, G-quadruplexes are associated with repeats, replication origins, and 5′ and 3′ UTRs. The Maculavirus vitis (Grapevine fleck virus) is the most G-quadruplex-rich organism found to date (≈23.8 putative G-quadruplexes per 1000 nt, 66% of its genome is covered by G-quadruplex-forming sequences) [210]. This extreme enrichment is the result of a very strong GC-skew (uneven asymmetry G versus C distribution between the two strands): the complementary negative (–) replication intermediate is extremely G-rich and forms long G-runs, which dramatically favors the G-quadruplex abundance. In contrast, G-quadruplexes tend to be detrimental to SARS-CoV-2 by creating steric hindrance for the viral RNA polymerase during replication and by blocking ribosome elongation [211]. At the same time, hairpin formation at the 5′ UTR promotes viral transcription [212]. Finally, Z-DNA acts as an innate immune cue via Zα-domain-containing proteins. Host cytosolic ZBP1 binds to viral Z-nucleic acids generated during replication or transcription and can trigger necroptosis and inflammatory signaling [213].
Non-B DNA motifs are abundant in repetitive regions in genomes and represent the cumulative outcomes of evolutionary processes, serving as molecular records of genomic history. Since non-B DNA structures can stimulate genetic instability, they can initiate genomic variation [21, 174, 214–216] and provide targets for selection and therefore are considered as drivers of genome plasticity and evolution [217–221]. For example, direct and inverted repeats in the terminal inverted repeats of common transposable elements can stimulate the formation of DSBs, which can lead to gap-filling and capturing of nearby sequences as fillers, or cause template switching [222]. Through these steps, transposable elements could amplify and spread themselves within genomes. Repetitive elements in centromeric and telomeric repeats can also stimulate the formation of DSBs and mediate recurrent chromosome fusions, drastically reducing the chromosome numbers; for example, from 2n = 70 in the ancestral karyotype to 2n = 6/7 in M. muntjak vaginalis [223]. Further, Z-DNA-forming sequences have been shown to stimulate recurrent deletions of a pelvic enhancer of the Pitx1 gene, resulting in many independently derived freshwater stickleback fish that adaptively lost pelvic hind fins in <15,000 years [224]. Structurally complex DNA sequences containing G-quadruplex, H-DNA, and hairpin motifs have rapidly expanded in eumetazoan genomes, indicating their potential evolutionary utility [225]. Their over-representation at promoters, enhancers, UTRs, and other functional elements positions mutation-prone DNA to be both a likely driver of genomic variation and often under selection constraints [226–228], is involved in aging, and a contributor to human diseases, including cancer.
Higher mutation rates at non-B DNA loci in germline and somatic contexts
Experimental systems have been instrumental in uncovering the mechanisms underlying the instability of non-B DNA-forming sequences. Genomic instability has been observed in vivo and in cultured cells for a number of non-B DNA-forming sequences, including inverted repeats [23], Z-DNA [24, 229], and H-DNA [22, 32]. In addition, non-B DNA motifs are detected more frequently in cancer cells than in normal cells [230]. While mutations can contribute to aging and many human diseases, the role of non-B DNA structures as drivers of these mutations through genomic instability is only partially understood [33, 231–233]. Systematic analysis of mutations from healthy and cancer genomes has provided critical evidence for the contribution of non-B DNA-forming sequences to germline and somatic mutagenesis [19, 31, 214, 216]. Base substitutions, insertions and deletions (indels), and structural variants/rearrangements have all been found enriched at non-B DNA loci [19, 31, 215, 231]. Different non-B DNA categories show higher enrichment for specific mutation types and under particular conditions or contexts.
Several non-B DNA structures can obstruct both the transcription and the replication machinery, leading to DNA and RNA polymerase stalling, fork collapse, and genomic instability [23, 150, 151, 234–240]. Beyond direct polymerase stalling, non-B DNA-forming sequences can also increase the probability of replication-transcription collisions when the two processes happen on the same DNA template. These conflicts can happen either head-on, when the replisome and the transcription complex approach one another from opposite directions, or co-directionally when a replication fork catches up to the transcription complex moving in the same direction, due to transcription stalling from a non-B DNA structure [124]. During replication, G-quadruplexes can form spontaneously on the leading-strand template within the newly unwound DNA region, directly behind the minichromosome maintenance helicase (MCM) and before nascent DNA strand synthesis (Fig. 4A). These G-quadruplexes can locally perturb replisome organization, leading to reduced DNA synthesis [241]. Similar replisome stalling has been observed in vitro for i-motifs, which can lead to fork collapse and DNA deletions [14, 242, 243]. R-loops can also impede transcription and replication [244–247]. For example, they can form behind RNA Pol II, where negative supercoiling unwinds the DNA duplex, allowing the 5′ end of the nascent RNA to hybridize with the template strand. In G-rich regions, this is accompanied by G-quadruplex formation in the non-template strand [51, 248] (Fig. 4B). An H-DNA-forming sequence present in the human MYC promoter has been found to stall replication forks, leading to the formation of DSBs [124]. Cruciform-forming inverted repeats likewise impede fork progression and can be processed by structure-specific nucleases (e.g., ERCC1–XPF). When cleaved, they can also lead to DSB formation and large deletions [249]. It is important to note that such outcomes are not strictly replication dependent. For example, H-DNA-, Z-DNA-, and cruciform-forming sequences can stimulate DSBs even in replication-deficient extracts, consistent with repair-driven mutagenesis that can occur in the presence or absence of replication [250].
Non-B DNA structure formation drives replication and transcription-associated genomic instability. (A) G-quadruplexes can cause replication fork stalling, leading to fork collapse and DSBs. (B) R-loops formed during transcription can block RNA Pol II progression and promote DNA breaks. (C) Short tandem repeats adopt slipped structures during replication, resulting in repeat expansions or contractions. (D) H-DNA and G-quadruplexes accumulate more oxidative DNA damage than B-DNA, which can interfere with repair processes, predisposing sequences to mutations. (E) Hairpin loops are susceptible to APOBEC3A-mediated cytosine deamination, generating hotspot mutations. (F) Cruciform structures form Holliday junction-like intermediates that undergo structure-specific cleavage, causing strand breaks and deletions.
Several specialized helicases have been found to resolve non-B DNA conformations, which, if left unresolved, can lead to DNA damage and mutagenesis [234, 235, 251–253]. To characterize such molecular interactions, X-ray crystallography [254, 255] and cryo-EM [256, 257] have been employed, revealing that they occur via selectively distinct interaction surfaces, which differ from those observed for duplex DNA. G-quadruplexes generated during replication are primarily resolved by FANCJ, BLM, and WRN, among others [258–261]. Evidence from model organisms suggests that in the absence of these helicases, G-quadruplexes can persist throughout the cell cycle, promoting genomic instability [252, 262]. Helicase depletion can increase fork stalling, DSBs, and rearrangements at G-rich loci [263]. R-loop resolution is also regulated by a network of specialized enzymes, including BRCA1, SETX, and TOP1, and their deficiency results in R-loop build-up [264–267]. Persistent R-loops can cause replication-transcription conflicts that generate mutations, DSBs, and large-scale chromosomal rearrangements [245, 268–271].
H-DNA is also found to have a broad interactome of triplex-associated proteins. DDX3X directly unwinds H-DNA, thereby preventing double-strand DNA breaks at H-DNA loci [272]. DHX9 has also been found to resolve H-DNA conformations in vivo and in vitro, and its absence leads to increased genomic instability and mutagenesis [273]. Both helicases also unwind G-quadruplexes. They resolve transcriptionally formed G-quadruplex structures that would otherwise lead to polymerase stalling [274, 275], as well as G-quadruplexes that stall ribosomes, which would in turn lead to depleted protein synthesis and cell senescence [276, 277]. These findings suggest that these helicases are not restricted to a single non-B DNA structure but are promiscuous to multiple non-B DNA types. In addition to causing replication stress, non-B DNA structures are frequently misrecognized by repair proteins as DNA lesions, thereby activating repair pathways, or as ssDNA resembling invading viral genomes, thereby triggering mutagenesis [250, 278, 279]. Such misprocessing can also interfere with the recognition and repair of DNA damage, leading to error-prone outcomes [280–282]. In addition, some non-B DNA-forming sequences can stimulate damage response pathways [124, 283, 284].
Replication fork stalling at unresolved non-B DNA structures can generate stretches of ssDNA coated with replication protein A (RPA), which can trigger ATR recruitment and Chk1 phosphorylation to activate cell cycle checkpoints [285]. When stalling progresses to fork collapse or the formation of DSBs, ATM-Chk2 signaling occurs, driving γH2AX formation and the formation of DNA damage foci [286]. In parallel, PARP1 acts as a sensor for both DNA damage and non-B DNA structures [287]. Upon binding to G-quadruplexes, R-loops, and cruciforms, PARP1 becomes catalytically active and synthesizes PAR chains, which recruit base excision repair proteins, single-strand break/DSB repair factors, and modulate chromatin structure [288–290]. Finally, G-quadruplex formation has been shown to recruit the Ku70/80 heterodimer to DSBs, which in turn recruits DNA-PKcs to the sites. The following phosphorylation of DNA-PKcs is crucial for the induction of DNA repair mechanisms [282]. Different DNA damage response pathways are associated with genomic instability across non-B DNA categories and are described in detail by Wang and Vasquez [6].
In addition to activating damage responses, repetitive non-B DNA sequences are intrinsically mutagenic through replication-dependent mechanisms. Direct and short tandem repeats can cause strand slippage events, resulting in frequent indels and substitutions [291] (Fig. 4C), a process that is intensified when mismatch repair is compromised. Unstable microsatellites undergo similar processes, producing a phenotype often observed in cancer [292]. Strand slippage events also occur at other non-B DNA types that coincide with repetitive sequences [19]. Trinucleotide repeat instability is a critical phenomenon underlying many neurological and other genetic disorders [232, 293, 294]. CAG repeats can form slipped DNA and hairpin structures, whose expansion or contraction depends on replication directionality [293]. Similarly, pentanucleotide repeats identified within introns have been associated with a variety of neurodegenerative diseases [295, 296]. Short tandem repeat sequences prone to forming slipped structures undergo expansions that can drive somatic instability, creating a random process of repeat length variation across tissues [297]. The structural instability and/or error-generating processing of these expanded repeats underscore the complex relationship between repeat-expansion dynamics and modulation of repair pathways. Finally, genome-wide analyses show that non-B DNA-forming sequences present in intergenic regions exhibit higher mutation densities than matched B-DNA controls [214, 228].
Non-B DNA-forming sequences are enriched at substitution sites, indel sites, and translocation breakpoints across cancer genomes [19, 31, 298], and have further been linked to copy number variation hotspots [299]. Some structure-forming sequences, such as G-quadruplexes, also coincide with oxidative DNA damage hotspots, particularly 8-oxo-guanine lesions leading to abasic sites and strand breaks [300, 301]. H-DNA-forming sequences also accumulate more oxidative lesions than B-DNA under conditions of oxidative stress [302] (Fig. 4D). In the human genome, thermodynamically stable inverted repeats with long arms, free of mismatches, are largely absent [303–306]. Experiments in which long inverted repeats are artificially introduced in eukaryotic cells show that these are genomic instability hotspots, which are lost within a few cell cycles [307]. Hairpin/cruciform-forming inverted repeats with long arms containing mismatches do exist, and they are linked to DSBs and chromosomal rearrangements [308–310]. In cancer genomes, inverted repeats are a mutational hotspot in part due to APOBEC enzyme off-target activity [216, 311, 312], but also via other mutagenic processing mechanisms, leading to the formation of DSBs and indels [19, 23] (Fig. 4E). It was also shown that functionally significant cancer “driver” mutation identification can be convoluted by the elevated mutation rates at pre-existing non-B DNA-forming sequences, which create recurrent mutational hotspots independent of selective pressure [19, 216]. Finally, inverted repeats are hotspots for DSBs and deletions in mammalian cells. Cruciforms formed at inverted repeat sequences resemble Holliday junctions in their four-way DNA structure, and can be recognized by resolvases (i.e., SLX1–SLX4, MUS81–EME1, GEN1) and nucleotide excision repair enzymes (e.g., XPF–ERCC1), resulting in genetic instability [23, 313] (Fig. 4F).
Additionally, stabilization of non-B DNA structures with ligands increases DNA damage responses and genomic instability, due to polymerase stalling, helicase inhibition, and aberrant repair. By stabilizing them with ligands, they act as roadblocks to stalling both RNA Pol II during transcription and Pol ε and δ during replication, leading to transcription-replication conflicts or replication fork collapse and finally the formation of DSBs [283, 314, 315]. The increased stability of G-quadruplexes in the presence of molecules such as PhenDC3 and TMPyP4 counteracts the activity of G-quadruplex-resolving helicases, enhancing previously mentioned events associated with genetic instability [283, 316]. G-quadruplex stabilization in telomeres has been shown to induce dissociation of the telomere-capping protein TRF2 and has also been found to prevent the formation of protective T-loop structures. This hinders repair in telomeres, and the effects can be expressed as sister telomere fusions and telomere loss [317, 318]. The effects of non-B DNA stabilizing molecules on genetic instability can be detected by immunofluorescence of γH2AX, p53BP1, pATM foci, ChIP-seq analysis of γH2AX genome-wide sites, and comet assays [269, 315].
While experimental studies provide mechanistic insights into key loci, computational analyses extend these observations to the genome-wide level. Whole-exome and whole-genome sequencing mutation datasets of healthy individuals and of tumor samples have offered valuable insights into the global impact of non-B DNA motifs. However, their interplay with various mutagen exposures and the presence/absence of DNA repair pathways remains only partially understood. Incorporating mutational signature analysis may help us better understand these complex interactions [311, 319] thus, future work in this direction is warranted
Biophysical determinants of non-B DNA-associated mutagenesis
As described previously, non-B DNA conformations contribute to localized increases in mutation rates. These effects are directly shaped by their biophysical and sequence properties, which affect the formation and stability of non-B DNA structures. Biophysical characteristics promoting more frequent or more stable non-B DNA formation are associated with heightened mutability, whereas features that destabilize them, including mismatches, reduce the observed mutation rates [19, 215, 216, 311, 320, 321]. G-quadruplexes with smaller loops form more readily, exhibiting higher mutation frequencies, as do inverted and direct repeats with short spacer lengths [19, 311, 320] (Fig. 5A). Using an APOBEC cytidine deamination activity assay, it was shown that biophysical properties of inverted repeats, including the spacer and arm lengths as well as the nucleotide composition, influence the levels of deamination activity. Tandem repeats become increasingly unstable as copy number grows [322, 323]. Mismatches within the repeat unit reduce mutability by destabilizing secondary structure formation [324–327] (Fig. 5A). Both repeat unit sequence and copy number are the primary determinants of tandem repeat formation across loci. Non-B DNA structures expose ssDNA, which is prone to damage and mutagenesis [19, 216, 311]. For instance, the exposed loops of G-quadruplexes [19, 174, 215] and of inverted repeats show an excess of substitutions [19, 216, 311], whereas Z-DNA accumulates mutations closer to the B-Z junction [215] (Fig. 5B). Clusters of non-B DNA structures result in even higher locally elevated mutation rates [315, 328, 329] (Fig. 5C). Depending on the cellular milieu, similar sequences can adopt different non-B DNA structures, with conditions that favor stabilization driving increased mutagenesis [269, 315, 329–335] (Fig. 5D). For example, Z-DNA forms under negative supercoiling caused by high transcription rates, inverted repeats generate transient hairpins during replication or repair, and K^+^ ions act as stabilizers for G-quadruplexes. Additionally, small-molecule G-quadruplex stabilizers are consistent drivers of increased DNA damage [269, 315].
Biophysical characteristics of non-B DNA structures directly influence associated genomic instability. (A) Structural features such as loop and spacer length modulate non-B DNA motif stability, with shorter loops or spacers exhibiting higher mutation rates. (B) Non-B DNA structures expose ssDNA that accumulates substitution mutations at loops of G-quadruplexes and inverted repeats and at B-Z junctions of Z-DNA. (C) Clusters of non-B DNA motifs generate locally elevated mutational burden. (D) The cellular environment directly influences the structural conformation of a nucleotide sequence. PQSs: Potential G-quadruplex sequences
Non-B DNA functional exemplars at cancer gene promoters
Studies focusing on cancer gene promoters have shown that non-B DNA-forming sequences coincide with mutational hotspots and can significantly change the expression levels of downstream genes. Multiple non-B DNA motifs were found in the promoter of the oncogene MYC, including G-quadruplex, i-motif, H-DNA, and Z-DNA conformations [148, 336–340]. Mutational disruption of the G-quadruplex leads to transcriptional loss, whereas small-molecule stabilization suppresses it [148, 340–342]. In the BCL2 promoter, multiple non-B DNA structures can be formed and have been experimentally tested for their regulatory roles [336, 343–345]. For example, G-quadruplexes formed in the P1 promoter are stabilized by oxidative lesions and can shift the equilibrium between B-DNA and G-quadruplex structures, potentially altering BCL2 transcription [346]. The KRAS promoter harbors G-quadruplex sites, which regulate expression levels and coincide with mutational hotspots [142, 347]. The TERT promoter is one of the most studied noncoding drivers across different cancer types [348]. Multiple mutational hotspots at this locus overlap with G-quadruplex loci, and the recurrent mutations identified disrupt both the G-quadruplex sequence and key transcription factor binding sites [349, 350]. Studies of the promoters mentioned above have shown that modulating the kinetics of non-B DNA conformation in these loci with small molecules can change the expression of downstream genes [333, 344, 351, 352], indicating their potential for drug discovery. Specifically, by manipulating the thermodynamic stability and interfering with the resolution of non-B DNA structures at target genes, new candidate drugs can be developed [353, 354]. Interestingly, recurrent repeat expansions have been observed across different cancer types, with most being cancer-specific, and a subset of these was shown to result in a dose-dependent decrease in cell proliferation [355].
Interpreting recurrence: drivers versus passenger mutations
Not all recurrent mutations in non-B DNA-forming sequences are consequential for cancer development. The prevailing dichotomy categorizes mutations as either drivers, which directly contribute to cancer development, or passengers, which are considered incidental byproducts of the genomic instability inherent to cancer cells [356]. Mutation recurrence has long been considered an indicator of positive selection. Understanding the determinants of the mutation rate variation in the human genome is pivotal for developing sensitive statistical models that can accurately detect selection in cancer.
Previous research has shown that recurrent mutations are more likely to overlap with non-B DNA sequences than expected by chance [19]. However, this might be driven by heightened genomic instability, selection, or both. Passenger mutation hotspots at inverted repeats in cancer-associated genes are often driven by APOBEC3A off-target mutagenesis [216]. For instance, recurrent mutations at the PLEKHS1 promoter, observed across multiple cancer types, occur within the spacer regions of inverted repeats and are attributed to APOBEC activity [321, 349, 357]. Nonetheless, their impact on gene expression remains inconclusive, as some studies report significant effects, while others do not [321, 349, 358]. Other inverted repeat mutation hotspots have also been characterized, with models developed to predict their mutability [216, 311]. These models account for the biophysical properties of the hairpin structure, including spacer and arm lengths as well as the nucleotide composition of each, which together influence the thermodynamic stability. In these cases, the heightened rate of mutagenesis is a more likely explanation than selection during cancer development.
Finally, the contribution of non-B DNA structures in driving human disease may be two-fold and context-dependent. First, noncanonical DNA conformations can have functional consequences, such as influencing gene expression, splicing, and translation. Secondly, their presence increases the likelihood of mutagenesis at a given site, which in certain cases can contribute to human disease and cancer development, while in others might have little or no effect. To investigate the impact of these mutations, examining the distribution of mutations within the sub-components of non-B DNA motifs and their surrounding regions, along with analyzing the biophysical properties of the motif and the associated mutational processes, can provide valuable insights. Further experiments are needed to elucidate their specific contributions and underlying mechanisms, which can differ from one case to another.
Non-B DNA as drivers of aging and tissue dysfunction
Non-B DNA loci might be pivotal for evolutionary adaptation, as they can accumulate mutations faster than the rest of the human genome and increase genetic variation at the population level. However, the same selection forces can act in somatic cells, resulting in clonality and tissue dysfunction. In somatic cells, the heightened genomic instability at non-B DNA loci can therefore drive human diseases and aging. Loss of the RecQ helicases, which drive premature aging syndromes, disrupts transcriptional regulation at G-quadruplex loci, linking G-quadruplex metabolism to age-associated gene expression changes [359]. WRN deficiency is also found to have a two-fold increase in mutational frequency, both for single base substitutions and deletions at H-DNA and Z-DNA-forming sequences [360].
In Alzheimer’s disease, hippocampal DNA from severely affected patients exhibits strong B-to-Z-DNA transitions. Z-DNA-forming sequences in the promoters of the APP, APOE, and Presenilin genes may alter their transcription and contribute to disease progression [361, 362]. In Huntington’s disease, long expansions of CAG repeats in the HTT gene progressively exceed a toxic threshold in vulnerable neurons, with expansion rates accelerating with age and driving neurodegeneration [363]. In amyotrophic lateral sclerosis and frontotemporal dementia, the GGGGCC hexanucleotide repeat expansion found in C9orf72 has been found to form G-quadruplexes and R-loops impeding RNA polymerase transcription, which results in transcriptional pausing and abortion [364].
Studies in model organisms have revealed tissue-specific, age-dependent increases in non-B DNA-associated mutagenesis. Age-related mutagenesis at H-DNA motifs from the human MYC gene increases in mouse spleen but not in testis [35]. Similarly, cruciform-induced genomic instability increased mutation frequencies in aging spleen and brain tissues of mice, compared to B DNA [365]. Additionally, H-DNA-induced mutagenesis has been found to increase with age in the mouse spleen and liver, and decrease in the brain [37]. In Drosophila melanogaster photoreceptor neurons, R-loop levels increased with age, and their accumulation across long, highly expressed genes disrupted gene expression and visual function. Notably, enhancing R-loop resolution via overexpression of RNase H1 or Top3β rescued visual decline [366].
Mitochondrial DNA (mtDNA) is also susceptible to non-B DNA-mediated instability. Deletions in mtDNA accumulate in aging tissues and can contribute to neurodegeneration, with hairpin- and cruciform-forming sequences significantly enriched at deletion breakpoints. G-quadruplex structures in mtDNA potently stall Pol γ in vitro, and analysis of human population cohorts reveals significant enrichment of variants within G-quadruplex-forming regions, suggesting these structures promote mutagenesis during replication [367–369].
Interestingly, non-B DNA-induced mutagenesis does not always increase with age, though non-B DNA-associated DSBs increase as well as apoptosis, which can also contribute to tissue dysfunction and aging [36]. Thus, overall, the faster accumulation of mutations at non-B DNA loci relative to B-DNA, and the increase in DSBs and apoptosis with age in somatic cells can account for a disproportionate contribution to aging. These emerging connections between non-B DNA-forming sequences and aging warrant further investigation. Population-level analyses of healthy aging individuals across tissues can help identify non-B DNA loci that predispose certain organs to age-related dysfunction. A deeper understanding of non-B DNA-mediated mutagenesis in aging may inform strategies to preserve genomic integrity and extend human healthspan.
Conclusions and future directions
Recent advances in long-read sequencing technologies have expanded our capacity to study non-B DNA-forming sequences. Long-read sequencing services from Oxford Nanopore Technologies [370, 371] and PacBio’s HiFi [372] allow for the generation of high-quality gapless reference genomes, as evidenced by the work performed by the Telomere-to-Telomere and Human Pangenome Reference consortia [373–375]. Non-B DNA-forming sequences are abundant in repetitive regions of the human genome [173, 201, 202], and human pangenome assemblies enable genome-wide analysis of non-B DNA motif distribution across previously missing repetitive regions [373, 375]. These technologies allow for the simultaneous readout of epigenetic modifications, which can be integrated with non-B DNA analysis, since formation kinetics and structure stability are directly influenced by methylation levels [187, 188, 376, 377]. Increased methylation levels destabilize G-quadruplexes [378], but aid Z-DNA formation [379]. Previously, elevated sequencing errors have been detected at non-B DNA-forming sequences [380, 381], because these sequences are often repetitive and deviate from the standard B-DNA conformation, suggesting that the higher mutation rates in non-B DNA regions may be attributed to sequencing artifacts. The new long-read sequencing technologies have significantly reduced sequencing error rates [382] and enable re-evaluation of these claims, revealing a consistent increase in mutation rates at G-quadruplex regions [174], consistent with previous experimental systems [22, 24, 383, 384].
These technological advances position the field to identify potential roles of non-B DNA-forming sequences in previously inaccessible genomic regions, particularly in the context of lineage-specific genome evolution and genetic diversity. Initial work in this area, utilizing high-quality genome assemblies of primates, has catalogued thousands of predicted non-B DNA motifs in repetitive regions [173, 201, 203].
Another key unanswered question is how non-B DNA structures contribute to somatic mutation accumulation and clonal expansion during aging. Ultra-deep, single-molecule sequencing technologies enable the detection of rare somatic mutations, providing the resolution to quantify low-frequency variants that accumulate in somatic cells during aging and in human diseases [385–389]. Paired with single–nuclei techniques for non-B DNA motif detection, they can reveal cell-to-cell heterogeneity [390]. Application of these technologies could reveal how noncanonical DNA structures contribute to mutational hotspots, clonal expansions, and age-associated genome instability, thereby uncovering links between DNA conformation, mutation accumulation, and the etiology of aging-related diseases.
In conclusion, non-B DNA structures play a dual role in human biology: they are key contributors to genetic variation and evolutionary adaptation, yet they also drive genomic instability that underpins somatic mutagenesis, tissue dysfunction, and human disease. The genomic instability found at non-B DNA loci, while promoting diversity and evolutionary potential, can also accelerate the accumulation of somatic mutations that compromise cellular function, ultimately contributing to aging and diseases (Fig. 6).
Noncanonical DNA structures act as functional elements but can also be sources of genomic instability. At cis-regulatory elements such as promoters, enhancers, and splice sites, non-B DNA motifs modulate gene expression and create localized mutational hotspots that foster genetic diversity and phenotypic heterogeneity. When unresolved, they cause DNA damage, disrupt homeostasis, promote aging, and contribute to the etiology of human diseases.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Watson JD, Crick FH. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature. 1953;171:737–8. 10.1038/171737 a 0.13054692 · doi ↗ · pubmed ↗
- 2Ghosh A, Bansal M. A glossary of DNA structures from A to Z. Acta Crystallogr D Biol Crystallogr. 2003;59:620–6. 10.1107/s 0907444903003251.12657780 · doi ↗ · pubmed ↗
- 3Kaushik M, Kaushik S, Roy K et al. A bouquet of DNA structures: emerging diversity. Biochem Biophys Rep. 2016;5:388–95. 10.1016/j.bbrep.2016.01.013.28955846 PMC 5600441 · doi ↗ · pubmed ↗
- 4Choi J, Majima T. Conformational changes of non-B DNA. Chem Soc Rev. 2011;40:5893–909. 10.1039/C 1CS 15153 C.21901191 · doi ↗ · pubmed ↗
- 5Georgakopoulos-Soares IC, Candace SY, Ahituv N et al. High-throughput techniques enable advances in the roles of DNA and RNA secondary structures in transcriptional and post-transcriptional gene regulation. Genome Biol. 2022;23:159. 10.1186/s 13059-022-02727-6.35851062 PMC 9290270 · doi ↗ · pubmed ↗
- 6Wang G, Vasquez KM. Dynamic alternative DNA structures in biology and disease. Nat Rev Genet. 2022;24:211–34. 10.1038/s 41576-022-00539-9.36316397 PMC 11634456 · doi ↗ · pubmed ↗
- 7Spiegel J, Adhikari S, Balasubramanian S. The structure and function of DNA G-quadruplexes. Trends Chem. 2020;2:123–36. 10.1016/j.trechm.2019.07.002.32923997 PMC 7472594 · doi ↗ · pubmed ↗
- 8Xie KT, Wang G, Thompson AC et al. DNA fragility in the parallel evolution of pelvic reduction in stickleback fish. Science. 2019;363:81–4. 10.1126/science.aan 1425.30606845 PMC 6677656 · doi ↗ · pubmed ↗