Currarino syndrome associated with an isolated 7q terminal deletion in Korea: a case report
Jin Hee Jung, Juiee Jeong, Sung Hyun Kim

TL;DR
A rare genetic disorder involving a deletion on chromosome 7 was identified in a Korean girl with Currarino syndrome symptoms.
Contribution
This is the first reported case in Korea of Currarino syndrome caused by an isolated terminal 7q deletion.
Findings
A 9-month-old girl presented with the complete Currarino syndrome triad.
Chromosomal microarray analysis identified a 12 Mb deletion at 7q35 to 7q36.3.
This case represents the first isolated 7q terminal deletion causing Currarino syndrome in Korea.
Abstract
The 7q terminal deletion syndrome is a rare genetic disorder caused by the deletion of the long arm of chromosome 7 between 7q32 and 7q36.3. It is characterized by various clinical symptoms, such as abnormal facial features and impaired mental and physical development. Currarino syndrome is defined by a triad of sacral bone defects, anorectal malformations, and presacral masses and is often associated with mutations in the MNX1 gene located at 7q36.3. Only a few cases of 7q terminal deletion syndrome have been reported in Korea. In one of these familial cases, Currarino syndrome was associated with a complex chromosomal rearrangement involving a 7q deletion and an 8q duplication. However, to our knowledge, cases of isolated 7q terminal deletions without other structural chromosomal abnormalities have not been described in the literature. We report the case of a 9-month-old girl who…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
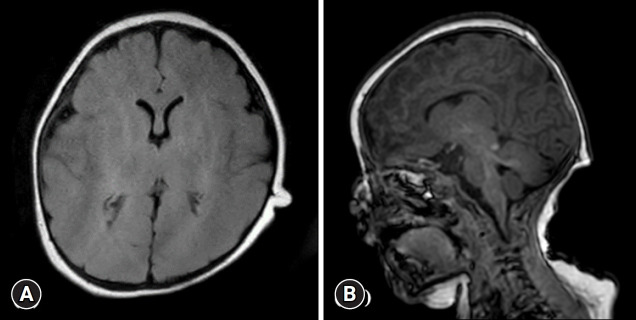 Figure 1
Figure 1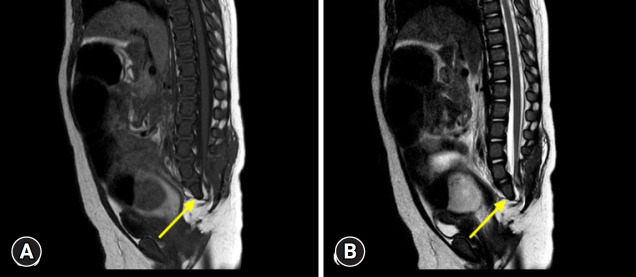 Figure 2
Figure 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCongenital gastrointestinal and neural anomalies · Congenital Ear and Nasal Anomalies · Genomic variations and chromosomal abnormalities
Introduction
The 7q terminal deletion syndrome was first characterized by Harris et al. [1] and encompasses a spectrum of clinical phenotypes caused by deletions in the long arm of chromosome 7 [1,2]. The common features of 7q terminal deletion syndrome are developmental delay, intellectual disability, and craniofacial abnormalities [1,3]. Although 7q36 deletions are often associated with holoprosencephaly because of Sonic Hedgehog (SHH) gene involvement [2], these deletions can also encompass the motor neuron and pancreas homeobox 1 (MNX1) gene, leading to distinct presentations. Only a few cases of isolated heterozygous chromosome 7q terminal deletions between 7q32 and 7q36.3 have been reported [3-7]. Most cases involve 7q36 microdeletions, and this genomic region contains approximately 40 Online Mendelian Inheritance in Man database genes, including SHH, engrailed-2 (EN2), enhancer of zeste 2 (EZH2), limb development membrane protein 1 (LMBR1), and MNX1. This genomic region may be dosage-sensitive, with key genes of clinical value for identifying and treating this disorder [4,8]. Variations and mutations within a gene can result in various phenotypic expressions, and MNX1 gene mutations have been linked to Currarino syndrome (CS) [9-11]. CS is defined as a clinical triad of presacral masses, anorectal malformations, and sacral bone deformity [12]. In Korea, previous reports of CS associated with 7q deletions involved complex rearrangements such as concurrent 8q duplications [7]. To the best of our knowledge, no case of an isolated 7q terminal deletion has been documented in Korean literature. Herein, we report the first Korean case of the CS triad caused by an isolated 7q terminal deletion.
Case
Ethics statement: This study was approved by the Institutional Review Board (IRB) of Bundang Jesaeng Hospital (IRB No: Bundang Jesaeng 2025-09-010). Informed consent for the publication of the patient’s clinical details and images was obtained from the patient’s guardian.
A 9-month-old girl was referred to the Department of Rehabilitation Medicine of our hospital on August 2, 2021, for intensive rehabilitation because of global developmental delay. She was born via cesarean section at 38 weeks and 3 days of gestation with a birth weight of 2.96 kg. Her family and perinatal histories were unremarkable.
At birth, she presented with multiple congenital anomalies, including micrognathia, microcephaly, frontal bossing, a short neck, a high-arched palate, and narrow nostrils, which prompted transfer to a tertiary hospital for further evaluation.
The initial workup, which included imaging studies, revealed a presacral mass, sacral agenesis, and rectal stenosis, confirming the presence of the CS triad. Chromosomal microarray analysis revealed a 7q35 to 7q36.3 (12 Mb) deletion. Additionally, a next-generation sequencing panel identified several variants of uncertain significance in other genes such as lysine demethylase 6A (KDM6A) and SET domain containing 2 (SETD2). Brain magnetic resonance imaging (MRI) performed on November 30, 2020, revealed no structural abnormalities (Fig. 1). During early infancy, the patient underwent a series of surgical interventions, including a laparoscopy-assisted colostomy in November 2020, colostomy repair in January 2021, and detethering of the lipomyelomeningocele on March 4, 2021. Lumbosacral spine MRI performed on April 2, 2021, after the surgeries, demonstrated a hypoplastic S2 vertebra and agenesis of the lower sacrum and coccyx (Fig. 2).
Her first developmental assessment at 7 months of age using the Bayley Scales of Infant Development-III showed no delay. However, the follow-up assessments at 17 and 22 months revealed developmental delays in all domains (Table 1). The Sequenced Language Scale for Infants score at 22 months of age indicated a language delay equivalent to that of a 9-month-old infant (Table 2).
The patient underwent continuous intensive outpatient rehabilitation including physical and occupational therapy. The follow-up evaluation at 37 months showed persistent delays across all areas, with only a slight improvement in the cognitive domain, suggesting that substantial catch-up had not occurred (Tables 1, 2).
Discussion
To the best of our knowledge, this is the first report in Korea of a patient with the complete CS triad caused by an isolated 7q terminal deletion. 7q terminal deletion syndrome was first characterized by Harris et al. [1] in 1977 and presents with characteristics such as growth retardation, intellectual disability, and atypical facial features [1,3]. A familial case of the Currarino triad associated with a complex chromosomal rearrangement involving 7q36.1 terminal deletion and 8q24.3 duplication was previously reported in Korea; however, a case with a pure 7q terminal deletion has not been described to our knowledge [7]. Here, we demonstrate how a specific genetic deletion leads to the clinical features of this rare syndrome.
The 7q36 region contains several dose-sensitive genes critical for embryonic development, such as SHH, EN2, EZH2, and MNX1 [8]. LMBR1, located at 7q36.3, contains a long-range enhancer of the SHH gene known as the zone of polarizing activity regulatory sequence (ZRS) [13]. Deletions of SHH or its regulatory elements, such as ZRS, are associated with holoprosencephaly [2,4]. However, the brain MRI of our patient was unremarkable, even though the deletion occurred in a critical region known to contain the SHH gene. This finding demonstrates the variable penetrance of holoprosencephaly as brain malformations do not manifest in all patients with deletions in this area [4].
Pathogenic variants of MNX1 are the major genetic causes of CS, a syndrome first linked to the 7q36 region [6,10]. MNX1 is a key transcription factor in the development of motor neurons and controls functions, such as movement and breathing [14]. However, MNX1 variants were not detected in our patient [9,11,15]. A comprehensive genetic review revealed that MNX1 mutations are identified in approximately 57% of patients with CS, with a lower detection rate in sporadic cases than in familial ones [9]. This genetic heterogeneity suggests that the phenotype does not rely solely on coding mutations. In cases of 7q terminal deletions, the most likely pathogenic mechanism is haploinsufficiency of MNX1, in which the loss of a single functional allele is insufficient for normal development [9,11]. Furthermore, 7q terminal deletions may disrupt critical regulatory elements within the 7q36 region that are essential for MNX1 expression, leading to the complete Currarino triad phenotype, even in the absence of coding variants.
Our patient met the diagnostic criteria for complete CS, as first described by Currarino et al. [12]. She presented with a sacral defect, anorectal malformation, and presacral mass [12,16]. The patient was diagnosed with a lipomyelomeningocele. This finding is consistent with CS because the presacral mass in this condition presents with various pathologies, such as lipomyelomeningocele, anterior meningocele, and teratoma [17]. The KDM6A and SETD2 variants that were also detected were considered incidental. Therefore, the complete CS triad observed in our patient was most directly attributable to haploinsufficiency of genes within the isolated 7q36 deletion. In contrast to a reported Korean case of a complex chromosomal rearrangement [7], our patient presented with an isolated 7q deletion, allowing for a more direct correlation between the genetic findings and development of the CS triad.
The patient experienced a persistent global developmental delay despite intensive rehabilitation, a common feature of 7q terminal deletion syndrome [1,8]. This highlights the need for long-term multidisciplinary follow-up involving continuous rehabilitation and developmental assessments.
In conclusion, we reported the first case of CS associated with an isolated 7q terminal deletion in Korea. This case expands the clinical spectrum of this rare condition and is genetically distinct from other 7q deletions previously reported in Korea [7,18].
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Harris EL Wappner RS Palmer CG Hall B Dinno N Seashore MR 7q deletion syndrome (7q 32 leads to 7qter)Clin Genet 197712233810.1111/j.1399-0004.1977.tb 00932.x 912940 · doi ↗ · pubmed ↗
- 2Frints SG Schoenmakers EF Smeets E Petit P Fryns JP De novo 7q 36 deletion: breakpoint analysis and types of holoprosencephaly Am J Med Genet 199875153810.1002/(sici)1096-8628(19980113)75:2<153::aid-ajmg 6>3.0.co;2-u 9450876 · doi ↗ · pubmed ↗
- 3Lukusa T Vermeesch JR Fryns JP De novo deletion 7q 36 resulting from a distal 7q/8q translocation: phenotypic expression and comparison to the literature Genet Couns 20051611515844773 · pubmed ↗
- 4Jackson CC Lefèvre-Utile A Guimier A Malan V Bruneau J Gessain A Kaposi sarcoma, oral malformations, mitral dysplasia, and scoliosis associated with 7q 34-q 36.3 heterozygous terminal deletion Am J Med Genet A 201717318586510.1002/ajmg.a.3827528488400 PMC 5680148 · doi ↗ · pubmed ↗
- 5Song JH Gonsalves Z Karlsen A Fiorentino D Dar PE Rabin-Havt S P 636: prenatal diagnosis of 7q, Xq/Yq deletion mosaicism in an alobar holoprosencephaly fetus: a case report and review of the literature Genet Med Open 2023110069210.1016/j.gimo.2023.100692 · doi ↗
- 6Lynch SA Bond PM Copp AJ Kirwan WO Nour S Balling R A gene for autosomal dominant sacral agenesis maps to the holoprosencephaly region at 7q 36Nat Genet 19951193510.1038/ng 0995-937550324 · doi ↗ · pubmed ↗
- 7Kwun Y Seo EJ Yoo HW Lee BS Kim KS Kim EA Phenotypic variability of a terminal 7q deletion/8q duplication in Korean siblings Ann Lab Med 2015355576010.3343/alm.2015.35.5.55726206699 PMC 4510515 · doi ↗ · pubmed ↗
- 8Fan LL Sheng Y Wang CY Li YL Liu JS Case report: congenital brain dysplasia, developmental delay and intellectual disability in a patient with a 7q 35-7q 36.3 deletion Front Genet 20211276100310.3389/fgene.2021.76100334925452 PMC 8671813 · doi ↗ · pubmed ↗