Integrase-On-Demand: bioprospecting integrases for targeted genomic insertion of genetic cargo
Hannah M McClain, Lillian C Lowrey, Laura B Quinto, Ellis L Torrance, Tomas R Gagliano, Farren J Isaacs, Joseph S Schoeniger, Kelly P Williams, Catherine M Mageeney

TL;DR
This paper introduces a new method called Integrase-On-Demand (IOD) to enable targeted gene insertion in a wide range of bacteria without needing to add special DNA sites.
Contribution
IOD identifies native integrase-attB pairs in prokaryotes, enabling site-specific integration without synthetic attB site introduction.
Findings
Diverse bacteria typically have multiple native attB sites usable for novel integrases.
IOD successfully mediated site-specific integration in Pseudomonas putida and Synechococcus elongatus.
Nonreplicating vectors were used to measure integration efficiency.
Abstract
Integrases serve as powerful biotechnology tools that catalyze recombination at specific DNA sequences (att sites) and facilitate chromosomal integration of gene cargos transferred into cells. Given that genomes often lack the attB integration sites recognized by frequently utilized integrases, integrase technology has largely been restricted to genetic engineering of model organisms into which attB sites can be synthetically introduced. To enable single-step site-specific integrase-mediated genome editing in a broad spectrum of prokaryotes, we have devised the Integrase-On-Demand (IOD) method. IOD systematically identifies integrases, within bacteria and archaea, that can integrate into available attB sites in any target prokaryote. Computational results show that diverse bacteria generally have multiple potentially useable native attB sites for novel integrases. We confirmed the…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
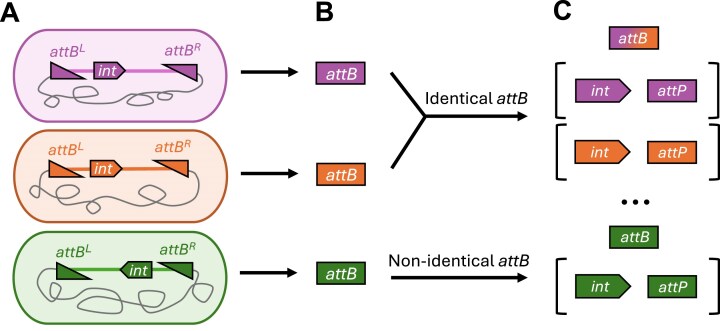 Figure 1
Figure 1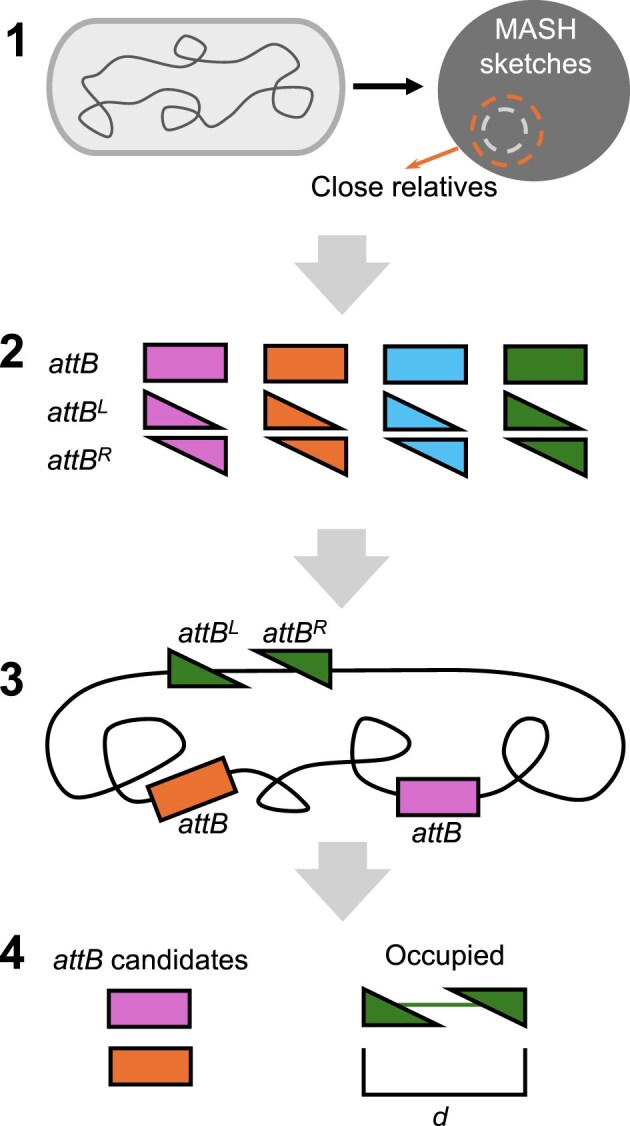 Figure 2
Figure 2 Figure 3
Figure 3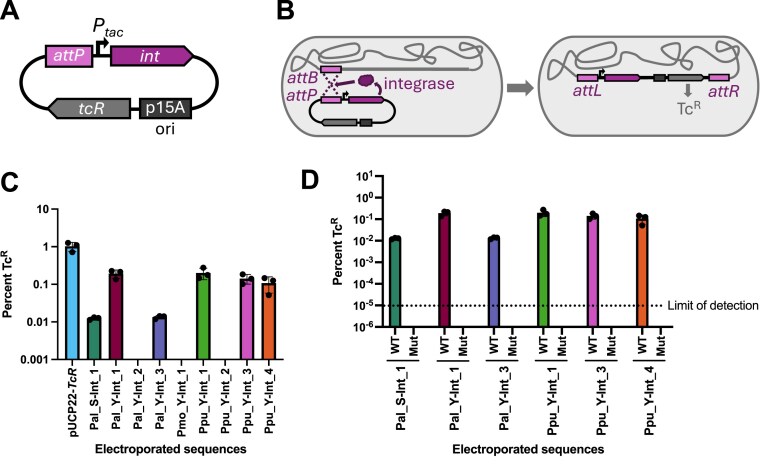 Figure 4
Figure 4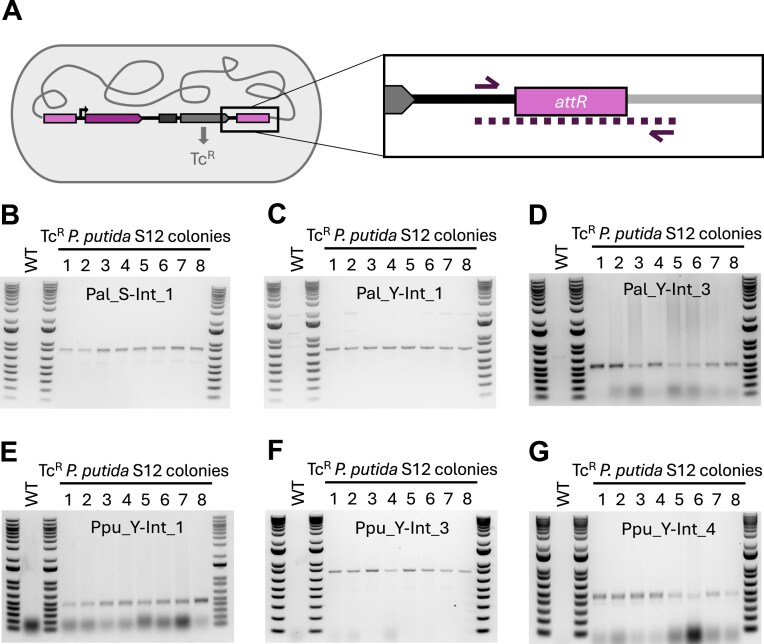 Figure 5
Figure 5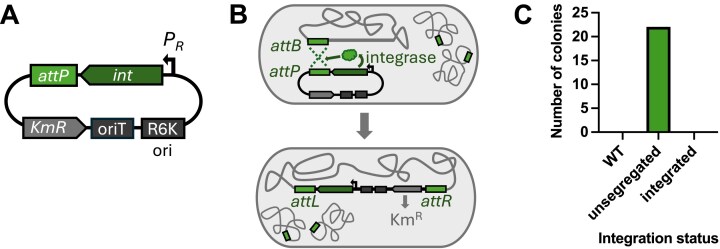 Figure 6
Figure 6| GTDB species | GCA |
|
|
| Occupied |
|---|---|---|---|---|---|
|
| 004359975 | 500 | 3 | 2 | 0 |
|
| 000011325 | 152 | 10 | 7 | 2 |
|
| 000025985 | 500 | 38 | 12 | 10 |
|
| 001457455 | 203 | 16 | 7 | 3 |
|
| 009759685 | 500 | 34 | 10 | 8 |
|
| 003482035 | 500 | 28 | 0 | 28 |
|
| 000359585 | 286 | 8 | 3 | 9 |
|
| 001457615 | 500 | 46 | 15 | 6 |
|
| 003054575 | 111 | 0 | 0 | 0 |
|
| 000495455 | 500 | 37 | 15 | 7 |
|
| 000143685 | 214 | 1 | 0 | 5 |
|
| 000012285 | 33 | 0 | 0 | 0 |
|
| 008639165 | 500 | 20 | 10 | 16 |
|
| 000012905 | 500 | 9 | 6 | 10 |
|
| 000007565 | 500 | 32 | 12 | 11 |
|
| 000013325 | 500 | 10 | 8 | 4 |
|
| 000025685 | 500 | 10 | 5 | 12 |
|
| 000008765 | 500 | 0 | 0 | 1 |
|
| 000621645 | 500 | 27 | 10 | 3 |
|
| 003610995 | 500 | 30 | 15 | 7 |
|
| 001043915 | 500 | 6 | 6 | 5 |
|
| 000817325 | 72 | 1 | 1 | 0 |
|
| 900234795 | 500 | 23 | 20 | 6 |
|
| 003697165 | 500 | 44 | 8 | 8 |
|
| 000392875 | 500 | 38 | 5 | 11 |
|
| 000742135 | 500 | 37 | 9 | 11 |
|
| 000009045 | 500 | 16 | 2 | 3 |
|
| 000011645 | 500 | 24 | 9 | 2 |
|
| 900478295 | 500 | 22 | 1 | 0 |
|
| 006716385 | 482 | 4 | 2 | 0 |
|
| 003315235 | 500 | 17 | 12 | 10 |
|
| 000009225 | 500 | 29 | 16 | 10 |
|
| 900187225 | 500 | 38 | 7 | 0 |
|
| 004798725 | 500 | 27 | 17 | 5 |
|
| 000021485 | 293 | 24 | 18 | 2 |
|
| 001457635 | 500 | 43 | 3 | 15 |
|
| 000755505 | 67 | 11 | 11 | 3 |
|
| 001027105 | 500 | 25 | 2 | 6 |
|
| 000006945 | 500 | 34 | 9 | 9 |
|
| 000195955 | 500 | 24 | 16 | 5 |
|
| 000008985 | 500 | 3 | 2 | 0 |
| Integrase name | IOD output integrase name | GI accession/coordinates |
|
|---|---|---|---|
| Pal_S-Int_1 | S-Int_05 038 | 5199833–5199794 | |
| Pal_Y-Int_1 | Y-BPP1Arm_00 722 | JAJSPR010000024.1/13977–1 + JAJSPR010000015.1/121491–103117 | 3624898–3624860 |
| Pal_Y-Int_2 | Y-P2Arm_04 004 | 4412428–4412467 | |
| Pal_Y-Int_3 | Y-P2Arm_04 082 | JAJSPR010000004.1/61465–7230 | 4394691–4394738 |
| Pmo_Y-Int_1 | Y-SXTArm_01 031 | 3480234–3480261 | |
| Ppu_Y-Int_1 | Y-P2Arm_04 153 | 1637680–1637745 | |
| Ppu_Y-Int_2 | Y-P2Arm_05 871 | 688725–688650 | |
| Ppu_Y-Int_3 | Y-SXTArm_00 656 | 1478260–1478293 | |
| Ppu_Y-Int_4 | Y-SXTArm_03 507 | 4394691–4394738 |
- —U.S. Department of Energy10.13039/100000015
- —Genomic Science Program, Office of Biological and Environmental Research
- —Secure Biosystems Design Science Focus Area, Intrinsic Control for Genome and Transcriptome Editing in Communities
- —InCoGenTEC
- —Sandia National Laboratories Laboratory Directed Research and Development
- —Office of Science10.13039/100006132
- —WDTS10.13039/100006210
- —Science Undergraduate Laboratory Internships Program
- —SULI
- —Department of Energy Office of Science BER
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCRISPR and Genetic Engineering · Enzyme Catalysis and Immobilization · Bacterial Genetics and Biotechnology
Introduction
Genome engineering enables sophisticated genetic and functional genomic studies in bacteria and microbiomes. Moreover, it drives advances in biotechnology applications, benefiting biomanufacturing [1], bioremediation [2], soil health and rhizosphere adaptation [3], and human health [4]. As biotechnology applications have diversified, and research into microbial ecosystem enabled by metagenomics has expanded, less well-studied, genetically and metabolically diverse microbes have elicited interest. These organisms often lack established genetic tools, or may be difficult to isolate or culture, which impedes the use of standard genetic tools for research and synthetic biology efforts to introduce novel or enhanced functions. Such concerns are particularly acute for applications such as, genetic engineering of specific members of microbial communities or microbiomes.
Mobile integrative genetic elements are delivered by phage particles or conjugation pili into bacterial or archaeal cells and integrate within the genome of the host as a genomic island (GI). Because of their foreign nature and mobility, GIs typically contain sequences which are not found in the host core genome but which are shared sporadic among strains of a species [5]. GIs often encode integrases that integrate their incoming DNA within the host chromosome. Integrases catalyze DNA recombination between attachment (att) sites in the mobile element (attP) and in the chromosome (attB) [6]. These attB sites are typically found within tRNA and tmRNA genes or intergenic sequences (56.6% of GIs from our recent TIGER database [7]), though many are found in protein-coding genes [5]. In many cases, Gis, unlike transposons, reconstitute the genes they insert within to preserve function [7–9]. However, it is possible that GI integration may lower host fitness. Recombination produces two hybrid att sites, attL and attR, that flank the GI within the chromosome, precisely defining its ends, and serving as substrates for a potential GI excision reaction that generally requires both the integrase and a Recombination Directionality Factor or excisionase (xis) gene [10]. Two unrelated integrase families exist, tyrosine and serine recombinases. While serine integrases are more commonly utilized in biotechnology, both families are capable of catalyzing integration, inversion, and excision of precisely defined regions of DNA.
Integrases provide a potential solution for efficiently introducing large genetic cassettes into a range of prokaryotes [11, 12]. They have many advantages over other methods of genetic engineering including insertion of large DNA payloads [12], scarless insertions [6], conservation of attB sites between related taxa [9], and site-specific insertion with minimal off-target activity [13]. Technologies have been developed to utilize integrases for genome engineering [14], including programmable addition via site-specific targeting elements (PASTE) [15], recombinase mediated cassette exchange (RMCE) [16], and serine recombinase-assisted genome engineering (SAGE) [17]. However, these methods typically require two transformation and selection steps. The first introduces non-native att sites as a ‘landing pad’ (i.e. integration site) for subsequent DNA cargo integration (e.g. using random, highly mutagenic transposon insertion) and the second delivers the integrase and associated cargo genes into the randomly placed att site. Transformation, especially for nonmodel bacteria and archaea, can be very inefficient, and in some scenarios, such as engineering a nonisolatable/nonculturable member of a microbiome, multiple transformation and selection steps are either not feasible or would simply destroy the community being studied [18–20].
To facilitate site-specific genomic insertion of large genetic cargo in a wide range of prokaryotes, including members of communities, we have developed the Integrase-On-Demand (IOD) method. Recent advances in GI discovery, enabled by our Islander and TIGER software, have greatly expanded the association of tyrosine and serine integrases with their precisely predicted att sites across hundreds of thousands of diverse prokaryotic genomes [20]. IOD starts with a computational pipeline that leverages these advances to identify prokaryotic serine and tyrosine integrases predicted to recognize attB sequences present within a user-defined bacterial or archaeal strain of interest. Here, we demonstrate that the IOD software can predict integrases and att sequences from diverse species. As proof of concept for the IOD method, we experimentally validated that nonreplicating vectors could insert site-specifically, efficiently, and stably into the chromosomes of two phylogenetically distant and biotechnologically relevant bacterial species: the gammaproteobacterium Pseudomonas putida [strains S12 (nine integrases) and KT2440 (three integrases)], and the cyanobacterium Synechococcus elongatus UTEX 2973 (one integrase).
Materials and methods
Development of IOD software
IOD software references a database of attB sequences and their corresponding integrase proteins to provide the user with a list of candidates for use in a target genome. Each attB/integrase pair is derived from a GI predicted by the TIGER/Islander programs [5, 8, 21]. These programs provide detailed information on the predicted GIs, such as the sequences of the recombinant sites, attB sequence, and identifiers for integrases encoded within the putative GI. The programs have been run on over 450,000 genomes, yielding 1,757,053 GIs containing integrases paired with their cognate att sites [7]. To build the reference data structure, the set of unique attB sequences of the predicted GIs was collected (Fig. 1).
Preprocessing steps for IOD Integrase and att pairs. (A) attB sites are extrapolated from TIGER/Islander predicted GIs. (B) attB-integrase pairs are built based on attB sequence identity. (C) At least one integrase and predicted GI is associated with each attB sequence.
Integrases theorized to be inactive due to truncation or lack of catalytic domains were excluded. To assess tyrosine integrase integrity, we compared these integrases to the 20 HMMs of the tyrosine recombinase database [22]. Putative tyrosine integrases with no HMM match, or that matched to the Xer or Integron HMMs (subfamilies that perform nonintegrase functions) were rejected. Furthermore, tyrosine integrases longer than 800 amino acids were rejected. Finally, a metric (∆∆HA) was developed to identify truncated proteins, calculating the number of amino acid positions of the HMM missing at the termini of the protein (∆H) and the number of amino acids at the termini of the protein exceeding the HMM hit (∆A), and taking ∆∆HA = ∆H - ∆A. We used a cutoff of ∆∆HA > 100, which passed 98.5% of the medium-sized tyrosine integrases (250–800 aa) and 8.9% of the short tyrosine integrases (<250 aa), to reject nominally truncated proteins. Lastly, serine integrases were rejected unless: (i) the integrase had the Pfam Resolvase domain followed by the Pfam Recombinase domain and was not too long (>800 amino acids) or too short (∆∆HA > 0); or (ii) our Tater software had previously classified it as S-Core_IS607 and it was not too long (>300 aa) or too short (∆∆HA > 0). As a result, the final list of attB sites queried by IOD is 182 734 unique sequences.
The IOD program features two modes (search and taxonomic) along with several command line options. Search mode can be invoked to search any sequence against the genome of interest. The user may provide specific attB sequences to search or use the entire de-duplicated attB database (182 734). If a match is found in the genome of interest, the attB site, as well as the sequence(s) of the cognate integrase(s), are returned to the user. Taxonomic mode searches for attB sites found in close relatives to the genome of interest. Users can modify the minimum number of attB sequences to collect in taxonomic mode prior to searching (default: n = 500), length of attB flanks from serine (default: sl = 16), or tyrosine (default: yl = 10) integrases, and number of threads for BLAST to use (default: threads = 1).
Taxonomic mode runs MASH [23], a program that uses a hash function to reduce genomic sequences to representative sketches. A sketch of the user-provided genome is created and compared to a pre-compiled database of 85 205 Genome Taxonomy Database (GTDB) representative species [24] and returns a list of genomes which have high similarity to the query. Starting with the best candidate species, the attB sequences and their respective integrase(s) from within the query species are collected, reaching out to other species or genera, as needed, to reach the desired minimum number (default: n = 500).
A query FASTA including each full reference attB, and its two half sites attB ^L^ (left flank and identity block of attB), and attB^R^ (identity block and right flank of attB) is prepared. The half sites are used to probe for GIs occupying attBs against a nucleotide BLAST database is produced from the user-provided genome [25] using the flag -task blastn. Hits are then rejected if they have (i)< 95% identity, (ii) more than one gap in the alignment, (iii) more than two mismatches, or (iv) any combination of two or more hits to the same full attB or its half sites in the genome. Hits rejected in the final filter are retained in a separate list of possible GI-occupied attBs. To characterize a GI-occupied site, the length of each reference GI utilizing the attB is collected from the database to determine which GI could be present. Each GI size is then compared to the distance between the attB^L^ and attB^R^ hits. If the attB halves map to different contigs, then the direction of the BLAST hit is used to determine which contig termini would be expected to be part of a GI. To be predicted as an occupied site, the normalized (for length of expected GI) difference must be <0.05:
\documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{upgreek} \usepackage{mathrsfs} \setlength{\oddsidemargin}{-69pt} \begin{document} \begin{eqnarray*} {\mathrm{ normalized}}\ {\mathrm{ difference}} = \frac{{{\mathrm{ expected}} - {\mathrm{ actual}}}}{{{\mathrm{ expected}} + {\mathrm{ actual}}}} \end{eqnarray*}\end{document}Following the categorization of all the BLAST hits, the candidates are passed through a final refining filter to remove questionable attBs. These are defined as attBs found within occupied sites, attBs with multiple copies of the ID-block (bioinformatic-based estimate of the crossover site) elsewhere in the genome, or attBs with single-nucleotide tandem repeats in the ID-block.
Despite deduplication of sequences in pre-processing, the list of candidates often contains overlapping attB sites. Shifted attB sites could be explained by mutations between closely related species that share a GI and/or the limit of precision in integration sites called by TIGER/Islander software [5, 8, 21]. The program bins all candidates by overlapping coordinates and attempts to select the highest quality sequence from each bin according to the best BLAST support score. If there are ties, then the site whose reference GI has the highest support will be selected. The best candidate attBs are reported in final_candidates.tsv in ranked order according to the best score value of the reference GI(s) from which the attB was sourced. Each candidate site reports an example reference GI according to the highest support. If an attB has more than one reference GI, the final column will be populated with the GI ID (GCA accession and gene ID) and integrase for each reference. All candidates, including questionable candidates removed by the refining filter and deduplicated sites are reported in attb_dupes.tsv. Additionally, all attB sites are reported with each associated reference GI in isles*.json*. Similarly, occupied sites are deduplicated by overlapping coordinates and reported in occupied.tsv along with the reference GI.
Software testing
Two sets of genomes were collected for study. The first set (Health) contained 41 genomes of health and biomanufacturing relevance. The second set (Diversity) contained 142 genomes each representing a different GTDB phylum with >4 genomes available.
Both genome sets were tested in taxonomic and search modes using default parameters (Table 1 and Supplementary Table 1). To simulate a typical user experience, the software was tested locally on a MacBook Air, M15,13. IOD was written in python (v3.12) and requires the external programs MASH (v2.3) (https://github.com/marbl/Mash) and the BLAST (v2.16) search algorithm to be available as a system-wide executable. The program can be run from any command line interface. Memory consumption was found to peak at 14.6 MB and 14.4 MB, for taxonomic and search mode, respectively (Supplementary Fig. 1). The software and README is available at https://github.com/sandialabs/Integrase-On-Demand.
Phylogenetic analysis
The protein sequence for each integrase predicted by the IOD software was obtained. Serine and tyrosine integrase sequences were treated separately. Protein sequences were aligned using MAFFT v7.526 [26] (mafft –auto) and maximum-likelihood trees were prepared using RAxML-NG v1.2.2 [27] (raxml-ng –all –msa tyr.afa –model LG + G8 + F –tree pars 10 –bs-trees 200). The resultant phylogenetic tree was viewed using FigTree v1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/) and rooted at the midpoint.
Protein alignments for catalytic residue confirmation
We used the same method as described above to create the aligned protein sequence separately for the serine integrases and tyrosine integrases. For the serine integrases we obtained the integrase reference sequences from Uniprot [28] using the following accession numbers: PhiC31 (Q9T221) and Bxb1 (Q9B086). We obtained Lambda (P03700) and Cre (P06956) as model tyrosine integrases. The aligned protein sequences were loaded into the SnapGene viewer to create the final figure and confirm catalytic residue positions.
GI prediction, induction, and recombination measurements
TIGER [5, 21] and Islander [8] were used to predict GIs in P. putida S12 (assembly: GCF_000495455.2).
Pseudomonas putida S12 was grown overnight at 30°C shaking in Luria broth (LB), diluted 3:100, and grown to an OD_600_ of 0.4 and 1 μg/ml of mitomycin C (MMC) was added. Samples (1 ml) were collected at 0, 1, 2, and 3 h post MMC addition. Cells were pelleted at 16000 × g for 2 min and genomic DNA was isolated using the Qiagen DNeasy blood and tissue kit (Qiagen, Venlo, Netherlands, 69506). Genomic DNA libraries were prepared using the Illumina DNA prep kit (Illumina, San Diego, CA, 20060060; Indexes: Illumina, San Diego, CA, 20018708). Libraries were pooled in eqi-molar fashion, and the final library was sequenced using an Illumina NextSeq 500 sequencer with the high-output 300-bp single-end read sequencing kit (Illumina, San Diego, CA, 20024908).
Sequencing reads were quality filtered using BBDuk (v36.11; http://jgi.doe.gov/data-and-tools/bb-tools/) with the following parameters specified: ktrim = r, k = 21, mink = 11, hdist = 1. The filtered reads were analyzed with Juxtaposer [29] to identify noncanonical reads compared to the P. putida S12 reference genome. The resultant juxtas.txt file was used to create a graph of recombinant reads by placing the identified noncanonical read into a single 500-bp bin across the genome.
Bacterial strains and culturing
Cyanobacterium S. elongatus UTEX 2973 was cultured under 100 or 200 µE white light at 38°C with 0.5% CO_2_ in 1× BG-11 medium (prepared from Sigma–Aldrich, Darmstadt, Germany, C3061-500ML). Donor (Ec100D pir + cloning strains) and helper (HB101 with conjugal plasmid pRL443) Escherichia coli strains for cyanobacterial conjugation were cultured at 30°C in LB Lennox medium (Sigma, L3022-6 × 1KG). NEB 5-alpha E. coli (NEB, Ipswich, MA, C2987H), NEB 5-alpha F’I^q^E. coli (NEB, Ipswich, MA, C2992I), and P. putida were cultured in LB medium (Sigma–Aldrich, Darmstadt, Germany, L3522) at 37°C (E. coli) or 30°C (P. putida). When applicable, media were supplemented with 100 µg/ml diaminopimelic acid (DAP; Fisher AAB2239106) (E. coli), 50 µg/ml carbenicillin (Sigma C1389-5G; E. coli), 34 µg/ml of chloramphenicol (Teknova, Hollister, CA, C0322) (E. coli), 50 µg/ml kanamycin sulfate (American Bio, AB01100-00010; S. elongatus), or tetracycline (Teknova, Hollister, CA, T3325) at 10 µg/ml (E. coli) or 25 µg/ml (P. putida).
Plasmid construction
To generate plasmids for integrase testing in P. putida, DNA sequences consisting of a 600 bp attP sequence (composed of the computationally determined att identity block flanked by GI DNA), a tac promoter, and the corresponding int were synthesized by Twist Biosciences (South San Francisco, CA). These sequences were then cloned into pACYC184 bearing additional I-SceI sites using NEBuilder HiFi Assembly mix (NEB, Ipswich, MA, E2621L). Integrase-testing plasmids were maintained with chloramphenicol selection in NEB 5-alpha F’I^q^E. coli. Plasmids encoding frameshifted integrases were generated through either error-prone DNA synthesis or using inverse polymerase chain reaction (PCR) to delete a nucleotide within the int sequence. Plasmids with mutant attP sequences contained 600 bp false attP sequences from att-flanking non-GI DNA.
To generate the positive control plasmid pUCP22-tcR, tcR (conferring tetracycline resistance) from pACYC184 was PCR amplified with Q5 DNA polymerase (NEB, Ipswich, MA, M0491S) and cloned into pUCP22 using restriction cloning (Eco53kI (NEB, Ipswich, MA, R0116S), XbaI (NEB, Ipswich, MA, R0145S), Monarch^®^ Spin PCR & DNA Cleanup Kit (NEB, Ipswich, MA, T1130L), Ligase (NEB, Ipswich, MA, M0202L). pUCP22-tcR was maintained in NEB 5-alpha E. coli with tetracycline selection. To verify all plasmid sequences, whole plasmid sequencing was performed by Plasmidsaurus (Louisville, KY) using Oxford Nanopore Technology with custom analysis and annotation.
Plasmids for integrase testing in S. elongatus UTEX 2973 were generated using Gibson assembly. Constituent fragments were amplified from template plasmids: a backbone previously used for integrase-mediated cargo delivery in S. elongatus (pCargo), the Sel_Y-Int_1 int, and an array of synthetic attP sites (Twist Biosciences). The assembly was performed with NEBuilder HiFi Assembly mix (NEB, Ipswich, MA, E2621L), and transformed into Lucigen EC100D pir + cells (BioSearch Technologies, Petaluma, CA, ECP09500). Clones were sequence-verified via whole-plasmid sequencing by Quintara Biosciences.
For expression of integrases in S. elongatus, the broad host range lambda phage P_R_ promoter, controlled by temperature-sensitive repressor cI857, was used; this promoter has been demonstrated for integrase expression across a diversity of bacteria [14]. Immediately downstream of P_R_ is the Bacteroides P_cepA_ promoter to further enhance broad-host applicability of the constructs [14]. The integrase was expressed with a strong RBS, and no terminator was explicitly included, based on cloning considerations. The nonreplicating vector on which the integrases (Sel_Y-Int_1 and the Bxb1 negative control) were delivered expresses a broad host range aph (kanamycin resistance) cassette, as well, under a native km^R^ promoter, the ermF promoter from IS4351, and the yeast P_TEF_ promoter which has demonstrated activity in E. coli [14]. The aph gene was followed by the yeast TEF1 terminator, and the strong bacterial rho-independent L3S2P21 terminator [30].
Integrase testing in P. putida S12 and KT2440
Electrocompetent P. putida cells were prepared by washing subcultures (OD_600_ = 1.0) in 4°C 10% glycerol solution thrice with centrifugation steps occurring at 3000 × g for 15 min. All experiments were conducted from the same batch of electrocompetent cells stored at −80°C. 25 fmol of purified plasmid were electroporated (2500 V, 25 mF, 200 W, 2 mm gap cuvettes) into 50 μl of electrocompetent P. putida S12 or KT2440 in triplicate. Cells were recovered in LB broth for 2 h at 30°C with aeration. Eight 10-fold serial dilutions of recovered cells were plated on LB with and without tetracycline. Plates were incubated for 18 h at 30°C and colony forming units (CFU) were measured. Percent Tc^R^ was calculated by dividing the number of tetracycline resistant CFU by the total CFU detected in the absence of Tc. Recombination efficiency was calculated by dividing the percent Tc^R^ following electroporation of integrase-testing plasmids by the percent Tc^R^ following electroporation of the positive control plasmid pUCP22-tcR.
To screen for correct insertion of plasmid sequences at the chromosomal attB site, colony PCR was conducted using OneTaq DNA polymerase (NEB, Ipswich, MA, M0486S) with one primer annealing to the plasmid DNA and its pair annealing to the P. putida genome adjacent to amplify across the attR site (Supplementary Table 4) (IDT, Coralville, IA). PCR products were run on an agarose gel for visualization with an Invitrogen 1 kb plus ladder (Invitrogen, Waltham, MA, 10787018) as a size standard.
Serial passaging
Tc^R^ colonies with integrated plasmids containing either Pal_S-Int_1 or Ppu_Y-Int_3 were cultured in LB medium at 30°C without tetracycline in triplicate. Every 24h, cultures were diluted to an OD_600_ of 0.1 in 2 ml fresh LB medium. This passage was repeated thrice, allowing for 4 days of growth in medium without tetracycline selection. Cultures were then diluted and plated to achieve single colonies on LB agar without antibiotics. A total of 72 colonies were then patched on LB agar containing tetracycline to identify the proportion of the population that had excised the integrated plasmid, and thus had lost Tc^R^.
To estimate the number of generations, the CFUs for cultures at an OD_600_ of 0.1 (0h CFU) and cultures 24 h later (24h CFU) were calculated. The number of generations (n) occurring over 24 h was calculated by n = [log(24h CFU) − log(0h CFU)]/log(2). This generation time was then multiplied by 4 to estimate the number of generations occurring over the 4-day period of serial passaging.
Whole genome sequencing for insertion-site specificity
Dense Tc^R^P. putida S12 growth (∼100 colonies) with integrated Pal_S-Int_1, Pal_Y-Int_1, Pal_Y-Int_3, Ppu_Y-Int_1, Ppu_Y-Int_3, and Ppu_Y-Int_4 test plasmids were independently resuspended in phosphate buffered saline. Genomic DNA was then extracted from each sample using the Qiagen Blood and Tissue kit. Genomic DNA was sequenced by Plasmidsaurus (Louisville, KY) with an average coverage of 64.6×. We analyzed the sequencing by using our Readstepper software to analyze the insertional boundaries (can be found in the following repository: https://github.com/sandialabs/TIGER).
Integrase testing in S. elongatus
To generate individual clones for screening integration outcomes in S. elongatus, recipient S. elongatus UTEX 2973 was inoculated from a colony and grown to saturation in 25 ml BG-11 in a flask. Donors bearing the Sel_Y-Int_1 test plasmid, and a control strain bearing the same plasmid with a Bxb1 integrase, and the HB101 helper bearing pRL443 [31] were inoculated from glycerol stocks and grown overnight with carbenicillin supplementation. The next day, cultures were washed once in LB and resuspended at an OD_600_ of 10. Each donor was mixed in a 1:1 volumetric ratio with helper. Recipient was diluted to OD_750_ = 3 in nonselective BG-11 media, mixed in a 1:1 volumetric ratio with the donor-helper mixture, and 100 μl of the mixture was spread on each of three autoclaved 25-mm HATF filters (Millipore, Darmstadt, Germany, HATF02500) on prewarmed BG-11 + 5% LB Lennox plates, for three replicates of each conjugation. Conjugations were incubated at 30°C in the dark at ambient CO_2_ overnight, then transferred to 38°C, 0.5% CO_2_, and 100 μE white light, for a total of 24 h of conjugation. Filters were then transferred to 5 ml liquid BG-11 and vortexed to lift cells. Lifted exconjugants were concentrated via centrifugation and spotted at various dilutions onto plates supplemented with kanamycin, wrapped in parafilm to retain moisture, and incubated at 38°C, 0.5% CO_2_, and 100 µE white light.
To test for integration of the Sel_Y-Int1 plasmid, kanamycin-resistant exconjugants were inoculated in 70 μl BG-11 media in a parafilm-wrapped 96-well V-bottom plate. Clones were grown for three days at 38°C, 0.5% CO_2_, and 200 μE white light, and then cultures were boiled at 65°C for 6 min followed by 95°C for 2 min. Boiled cells were subsequently screened for integration via PCR using Kapa 2G Fast master mix (Roche, Basel, Switzerland, KK5609) with primer pair OLQ1039/OLQ257 (targeting the locus with integrated plasmid) and OLQ1039/OLQ1038 (targeting the wild-type locus), cycling for 35 cycles with a 57°C annealing step and a 20s extension (Supplementary Table 4). Products were analyzed on a 1% agarose gel to determine presence or absence of integrated plasmid.
To test integration efficiency in S. elongatus (Supplementary Fig. 9), conjugations were performed identically, with the following modification: Three donor colonies for each donor (Sel_Y-int_1 and Bxb1) were inoculated from streakouts on LB plates supplemented with carbenicillin. After the 24-h conjugations, cells were lifted from filters in 5 ml BG-11 medium, spun to concentrate, and resuspended in in 70 μl BG-11. These were serially diluted 15:60 in BG-11, and spotted in 5 μl spots on 30-ml BG-11 or BG-11 + kanamycin plates.
Statistical analyses
All integrase testing in P. putida was conducted using three biological replicates. When present, error bars on graphs denote standard deviation. For S. elongatus integrase efficiency testing, data was collected in biological triplicate, and to generate colonies to assay clonal integration outcomes, conjugations were performed in technical triplicate, and at least seven colonies were picked from each replicate.
Results
Development of IOD software
The IOD software identifies attB sites within bacterial genomes of interest to facilitate integrase-based integration of genetic cargo. We developed an attB database, derived from genomic data of GIs previously identified in our TIGER/Islander GI database of predicted GIs [7] resulting in a nonredundant list of 182 734 attB and integrase pairs. The IOD software can act on a query genome sequence in two modes: taxonomy mode identifies attB sites from close relative GIs or search mode which can search for attBs from the entire de-duplicated database (Fig. 2; see the ‘Materials and methods’ section).
Workflow for IOD software in taxonomic mode. (1) MASH is used to determine close relatives of the input genome available in the IOD database. (2) A query list of attBs and half attBs (see the ‘Materials and methods’ section; represented as triangles) are used as a query against the BLASTN database of the genome of interest. (3) A series of filters locates candidate attB sites and collapses overlapping matches. (4) GI-occupied attBs (identified by distance between hits to left and right attB halves) are rejected and the best candidate attB sites in the genome are reported separately from the duplicate sites.
Taxonomy mode is preferred for its computational efficiency (Supplementary Fig. 1) and biological basis. Biologically, the advantages are two-fold. (i) Taxonomically close relatives of the target bacterium are likely to exhibit similar cellular physiology and produce the same suite of integration host factors required for optimal integrase activity [32–35]. Integrase-att pairs output by IOD in taxonomy mode all function within relatives, as they have been able to insert the GI from which our sequences were sourced, increasing the likelihood that the integrase will function in the target strain. (ii) Integration events frequently interrupt coding regions, often tRNA genes [5, 8, 36]. Many attP sites have evolved to complete the gene that their GI is inserting within to maintain appropriate expression following integration.
To demonstrate broad potential utility of IOD for genome editing in phylogenetically diverse hosts, we have applied the IOD software to 183 genomes from a broad phylogenetic range of bacterial and archaeal species (Table 1 and Supplementary Table 1). Strains in Table 1 include many relevant for biomanufacturing or medical applications and were typically found to contain numerous candidate attB sites identified using IOD in taxonomy mode, with only three exceptions (Table 1). However, most other species we tested did not have candidate attBs predicted in taxonomy mode (97/142; Supplementary Table 1). For species in many phyla, especially those known only from environmental metagenomics, this is due to a lack of sequenced genomes from close relatives, resulting in too few attB search sequences. However, when running in search mode using all attB sequences in our database, only one strain out of the 183 genomes tested lacked an attB candidate; the total number of available attBs found in taxonomic mode was 234, rising to 1295 in search mode (Supplementary Table 1). There are no obvious trends as to which organisms are sources of the additional candidate attB sites found using search mode. In the cases we examined closely, source organisms span the entire phylogenetic tree with some crossing domains (e.g. integrase source is archaea and target was bacteria), further highlighting the utility of querying a large database for putative attB sites. These results indicate IOD software will find attB/integrase pairs for most bacteria and identify potential genomic locations for large DNA payload integration.
Using the 183 genomes representing diverse bacterial and archaeal phyla (Table 1 and Supplementary Table 1), we benchmarked performance speed. The average runtime per genome for both test sets was 21.93 ± 1.320 (20.61–23.25) seconds, which was not measurably influenced by genome size (0.37–16 Mbp) (Supplementary Fig. 2A). Runtime is primarily dependent on the number of attB sequences used to query against the genome of interest (Supplementary Fig. 2B). The maximum number of reference attB sequences collected from the closest relatives (based on phylogenomic comparison) of the input genome is 500 for taxonomic mode (run with default setting). However, there may be fewer attB sequences in the query depending on the species (see the ‘Materials and methods’ section), which results in a shorter runtime (e.g. S. elongatus). If the user runs the program in search mode without target sequences, the default database size is 182 734 (our entire attB database). The shortest allowable attB sequence in the database is 22 nucleotides (with default parameters). This was chosen to reduce false positives: given the probability of finding a random k-mer in a genome of length L is 2 × L × 4^−k^, the expected discovery rate for a random 22-mer in the P. putida S12 genome is ∼3.67 × 10^−7^, and for at least one of 500 22-mers, 1.84 × 10^−4^. Benchmarking results emphasize the value of taxonomic mode for attB sequence identification given the low expected discovery rate and the correlation between runtime and number of query attB sequences. However, if computational resources or time are not of concern, search mode may be preferable to increase to odds of identifying an integrase/attB pair for a target prokaryote.
Prediction and computational analysis of IOD integrases in P. putida
Pseudomonas putida is a Gram-negative soil saprophytic bacterium that has been embraced as a biomanufacturing and synthetic biology chassis due to its hardiness, genetic tractability, and metabolic versatility [37]. To refine our software predictions, we focused on the well-established laboratory strain P. putida S12. We predicted 37 candidate attB sites using taxonomy mode (Supplementary Table 2) and cross-referenced with the TIGER/Islander predicted GIs (Supplementary Table 3) to verify that IOD differentiated attB sites from attL/attR sites (which already contain a GI and thus should not be provided to the user as available candidate attB sites). Although GI-occupied attBs can be utilized, the pre-existing integrases and/or excisionases may destabilize a new insert that uses its att sites.
The IOD software attempts to remove any spurious integrase/attB pairs as part of the upstream processing steps; however, additional verification may be necessary. In our initial test set for P. putida S12, we determined that the predicted Pmo_Y-Int_1 integrase was ∼125 aa shorter than expected [our predicted integrases had an average length of 393 aa, and the model tyrosine integrases are 356 aa (Lambda) and 343 aa (Cre)]. We developed and implemented a filter to remove short integrases (see the ‘Materials and methods’ section), filtering out 315 597 of 923 744 unique tyrosine integrase sequences and 164 205 of 353 354 unique serine integrase sequences (mainly lacking the recombinase domain) from our initial integrase/attB site list.
To ensure we were testing functionally distinct integrases, we constructed two phylogenetic trees to determine the relationship between our IOD predictions for P. putida S12 (Fig. 3). All predicted integrases were unique, except for three (Y-STXArm_01597, Y-STXArm_01 983, Y-STXArm_02 423). We chose to experimentally validate nine integrases from across the two trees (Table 2 and Fig. 3).
Phylogenetic tree of IOD predicted integrases for P. putida S12. Protein sequences were aligned to create a maximum likelihood tree for the (A) Serine integrases (S-) and (B) Tyrosine integrases (Y-). Based on results from experimental validation (Fig. 4), functional integrases are colored green and marked with a check and failed integrases are red colored with an X. Branch support values are ≥95% except for those marked with a triangle (50%–94%) or a circle (<50%).
We evaluated the integrase protein sequences to determine whether the catalytic serine or tyrosine residues were present. Previous work on phiC31 and Bxb1 serine integrases identified essential residues for enzymatic activity; these include a conserved N-terminal domain [38], the catalytic serine (S10 in Bxb1 [39] and S12 in phiC31 [40]), and conserved residues found in the active pocket required for catalytic activity (R8, Q22, D81, R82, L83, R85 in Bxb1; R10, Q26, R93, R96 in phiC31) [41]. We confirmed that in all predicted serine integrases for P. putida S12, the catalytic serine residue was present, the N-terminal domain showed a high level of conservation, and the active pocket residues were fully conserved (Supplementary Fig. 3).
In tyrosine integrases, the C-terminal catalytic domain is characterized by the crucial Tyr and the Arg-His-Arg catalytic triad [42]. In all predicted tyrosine integrases, we identified the catalytic tyrosine (Supplementary Fig. 4). We observed a few mutations in the catalytic triad in the integrases in this dataset. In two integrases, the first arginine residue of the catalytic triad is mutated to a serine residue. In two integrases that were candidates for experimental validation (Pal_Y-Int_1 and Pal_Y-Int_2), the second arginine is mutated to a lysine. We also see a large clade of Y-BPP1 integrases (which includes Pal_Y-Int_3) missing the H308/H289 residue from the catalytic triad. We expect this to be a novel clade of integrases.
The lack of active recombination is an important criterion for selecting attBs for targeted integration because spurious excision or recombination events can reduce the stability of or damage the inserted genetic cargo. To test which predicted attB sites in P. putida S12 were optimal candidates for experimental validation of IOD, we treated P. putida S12 with MMC, a nonspecific DNA damaging agent, which often induces GI excision or other recombination events (potentially caused by transposons, homologous recombination, or nonhomologous end joining [43, 44]). Deep sequencing revealed that none of the attB sites that were selected for experimental validation in P. putida S12 were actively undergoing recombination (Supplementary Fig. 5). While multiple regions in the P. putida S12 genome have sequencing reads indicating recombination above background levels, none of these correspond to predicted GIs (Supplementary Table 3), surprisingly, suggesting that the GIs in this strain are not excised upon MMC treatment.
Experimental validation of predicted integrases for P. putida S12
We constructed tetracycline resistance-conferring, nonreplicating vectors containing 600 bp attP sequences (Supplementary Fig. 6) and the tac promoter driving expression of the cognate integrase (int) gene (Fig. 4A and Supplementary Table 5). Because the vector does not replicate in Pseudomonas, plasmids that bear sequences encoding a nonfunctional integrase will be unable to recombine into the chromosome and thus the plasmid, and the conferred tetracycline resistance, will be diluted out of the population. If the int sequence encodes a functional integrase and the predicted attB and attP are correct, the protein should mediate recombination between the plasmid attP sequence and the chromosomal attB sequence, stably conferring tetracycline resistance (Tc^R^) (Fig. 4B).
Using a nonreplicating vector-based approach, 6/9 tested integrase and attP pairs mediate recombination in an integrase and attP sequence-specific manner. (A) Diagram of an integrase testing plasmid. The testing plasmids bear the integrase gene (int) driven by a Ptac promoter, the 600 bp cognate attP sequence, a tetracycline resistance-conferring gene (tcR), and an origin that will not replicate in Pseudomonas (p15A ori). (B) Schematic of integrase-mediated recombination between attP and attB sites that integrates the integrase testing plasmid sequences into the chromosome, conferring stable tetracycline resistance. (C) Percent of the population with tetracycline resistance (TcR) following electroporation of 25 fmol of an integrase test plasmid bearing the int and cognate attP of the listed integrase or a positive control, replicating plasmid, pUCP22-TcR. The limit of detection was 10−5% TcR cells. (D) Percent of the population with tetracycline resistance (TcR) following electroporation of 25 fmol of wildtype (WT) a functional integrase test plasmid or (Mut) a plasmid containing either an incorrect attP sequence (Pal_Y-Int_1, Ppu_Y-Int_1, Ppu_Y-Int_3, Ppu_Y-Int_4) or a frameshifted int (Pal_S-Int_1, Pal_Y-Int_3). Values reported in panels (C) and (D) are raw numbers and not normalized.
Electroporation of plasmids bearing integrases Pal_S-Int_1, Pal_Y-Int_1, Pal_Y-Int_3, Ppu_Y-Int_1, Ppu_Y-Int_3, and Ppu_Y-Int_4 yielded an average of 0.01%, 0.19%, 0.01%, 0.20%, 0.14%, and 0.11%, Tc^R^ cells respectively (calculated by Tc^R^ CFU/total CFU) (Fig. 4C). Therefore, these six integrases and attP pairs identified by the IOD software are capable of facilitating DNA recombination in P. putida S12. As expected, the heavily truncated Pmo_Y-Int_1 failed to yield Tc^R^ colonies, as did two additional integrases, Pal_Y-Int_2, and Ppu_Y-Int_2, indicating that these three predicted integrases could not mediate recombination between the identified att sites in our assay (Fig. 4C).
Because acquiring stable Tc^R^ relies on successful electroporation followed by integrase-mediated recombination, to calculate integration efficiency, we must account for electroporation efficiency. To assess the rate of plasmid uptake through electroporation for our population, we generated pUCP22-tcR, a tetracycline-resistance conferring, replicating plasmid that is similar in size to the P. putida S12 integrase-testing plasmids. Electroporation of 25 fmol of this plasmid yielded a population with 1.02% Tc^R^ cells, indicating that in our assay, electroporation will deliver a plasmid of nearly 6100 kb to 1/100 cells in the population (Fig. 4C). At this electroporation efficiency, integrases Pal_S-Int_1, Pal_Y-Int_1, Pal_Y-Int_3, Ppu_Y-Int_1, Ppu_Y-Int_3, and Ppu_Y-Int_4 facilitated recombination in an average of 1.2%, 18.7%, 1.3%, 19.4%, 13.8%, and 10.5% of the cells that received the plasmid (Supplementary Fig. 7).
To verify that plasmid insertion into the chromosome is dependent on integrase-mediated recombination at attB sites and not mediated by other modes of recombination, we created additional mutant plasmids with either (i) single nucleotide deletions in the int sequence to produce frameshifted, nonfunctional integrase proteins or (ii) false attP sequences (see the ‘Materials and methods’ section). We observed that mutated versions of functional integrase/attP pairs did not generate Tc^R^ cells, and thus, we conclude that recombination is dependent on integrase activity and attP site sequence specificity (Fig. 4D). To further verify that insertion is attB sequence-specific, we PCR amplified attR regions using a primer annealing to the plasmid backbone and a primer annealing to the chromosome (Fig. 5A). For each integrase that mediated recombination, 8/8 screened Tc^R^ colonies yielded PCR products of the size expected for integration within the attB site, and no amplification was observed in the wild-type P. putida S12 negative control (Fig. 5B–G). To further probe for off-site genomic insertions, we conducted whole genome sequencing on mixed populations of Tc^R^ cells (∼100 colonies growing on a tetracycline-supplemented plate), following independent electroporation of the six functional integrase test plasmids. Whole genome sequencing for all tested integrases indicated that integration occurred at the predicted attB site, forming the expected attR and attL sites, with no evidence of off-target insertions. Together, these results indicate that the tested integrases are mediating recombination in an attB site-specific manner. Interestingly, tandem copies of inserted plasmids were present in a small amount of sequencing reads mapping to the test plasmid (1%) within half of the samples. These duplicate copies could be created by integration at a reconstituted attB site following successful integration of the first copy or by homologous recombination between the >6 kb of sequence homology shared between an integrated construct and an incoming plasmid.
Functional integrases mediate site-specific recombination between predicted attB sites. (A) To verify recombination occurs at the predicted chromosomal attB and plasmid attP sites, DNA from attR-containing junctions were PCR amplified using a primer annealing to chromosomal DNA and a primer annealing to plasmid DNA. (B–G) PCR products using wild-type P. putida S12 (WT) or TcRP. putida S12 colonies post electroporation of int genes encoding (B) Pal_S-Int_1, (C) Pal_Y-Int_1, (D) Pal_Y-Int_3, (E) Ppu_Y-Int_1, (F) Pal_S-Int_1, (G) Ppu_Y-Int_4, and cognate attP containing test plasmids.
Integrase-inserted gene cargo should remain stably integrated when introduced unless cognate excisionases are present to allow the reverse reaction to occur or there are other sources of genome instability. To test the stability of our integrated constructs, we serially passaged P. putida S12 with two integrated cargos (Pal_S-Int_1 and Ppu_Y-Int_3) in antibiotic-free medium every 24 h for 4 days. By comparing growth on LB with and without tetracycline supplementation, we observed that the Pal_S-Int_1 test cargo remained stably integrated in the genome (0% Tc^S^), while the Ppu_Y-Int_3 test cargo excised in a small proportion of cells (1.39% Tc^S^) over approximately 26 generations. Excision may be due to induction of a cross-reactive excisionase (five putative excisionases and 22 integrases are annotated in P. putida S12), or if the integrase is capable itself of catalyzing excision (i.e. it is bidirectional), or other processes such as endogenous recombination. Depending on the cause of mobility, one can improve stability by utilizing integrases experimentally verified to mediate stable insertion or maintaining (e.g. antibiotic) selection pressure. Cross-reacting excisionases already present in the target genome can be addressed by selecting an alternative integrase or curing the strain of the offending interfering GI. Bidirectional integrases can also be identified experimentally and excluded, or be tamed by providing the int on a separate nonintegrating plasmid, or genetically inactivating the int once the construct is integrated.
We also used IOD to identify integrase and attB pairs for P. putida strain KT2440 (Supplementary Table 2), which is used more frequently than P. putida S12 in biomanufacturing applications [37]. We observed that three of the six functional integrases tested in S12 were also predicted by the IOD software for KT2440 (Pal_Y-Int_1, Ppu_Y-Int_1, and Ppu_Y-Int_3; Supplementary Table S2). Using the same integrase test plasmids and assay, we demonstrated that all three integrases facilitated recombination in a sequence-specific manner, as evident from their generation of stably Tc^R^ colonies at 1.14%, 2.43%, and 2.39% of the total population respectively, and an absence of colonies for the controls lacking integrase activity (Supplementary Fig. 8A and B).
Experimental validation of a predicted integrase in S. elongatus
To further demonstrate the potential utility of the IOD method, we sought to test a predicted integrase and attP pair for site-directed chromosomal integration in a cyanobacterial strain, S. elongatus UTEX 2973. Many cyanobacteria are notoriously difficult to engineer, due to their high copy number of chromosomes per cell [45, 46], and slow chromosome segregation rates, so tools for efficient genomic integration are valuable. We applied our IOD software to S. elongatus UTEX 2973 in taxonomy mode and discovered a single integrase and attB pair (Supplementary Table 2). To test this integrase in our target strain, we conjugated S. elongatus with a nonreplicating vector bearing the lambda phage pR promoter-driven int encoding the Sel_Y-Int_1 integrase, attP site, and a kanamycin resistance marker to select for integrants (Fig. 6A and B). To control for nonspecific integration, we also conjugated the same construct with the Sel_Y-int_1 int replaced with the Bxb1 int, whose attB site is not found in the S. elongatus genome. Exconjugants from Sel_Y-int_1 grew on selective media, while the Bxb1 exconjugants did not (Supplementary Fig. 9), indicating that survival of the Sel_Y-int_1 transformants was mediated by Sel_Y-int_1 activity towards its native attB site. Spotting of serial dilutions of exconjugants suggested that integration efficiencies were ∼1%, but due to the density dependence of the cyanobacterial growth on selective media, precise integration efficiencies could not be ascertained (Supplementary Fig. 9). To confirm that selected exconjugants had undergone integration of the test plasmid, we screened kanamycin-resistant (Km^R^) colonies for integration at the native attB site via PCR. Because S. elongatus bears numerous chromosome copies, successful Sel_Y-Int_1-mediated recombination may occur at one or multiple attB loci in a single cell, resulting in a colony with a mixture of integrated and wild-type chromosome copies (“unsegregated”). All tested colonies demonstrated integration in some chromosome copies, and in most colonies, WT bands were very faint, suggesting almost complete segregation at the locus (Fig. 6C and Supplementary Fig. 10).
Testing Sel_Y-Int_1 integration at native cyanobacterial S. elongatus attB site. (A) Schematic of the kanamycin resistance-conferring, nonreplicating plasmid bearing an R6K ori, origin of transfer for conjugation (oriT), the Sel_Y-Int_1-encoding int driven by the lambda phage pR promoter, and the cognate attP site. (B) Schematic of integrase-mediated recombination between the plasmid attP site and the many copies of the chromosomal attB site, which leads to stable kanamycin resistance. (C) KmR clones obtained were assayed for integration via PCR of the attB site. “Integrated” clones would have yielded only a band detecting the integrated plasmid, whereas “unsegregated” clones displayed both wild-type “WT” and integrated loci. WT clones would have displayed amplification for only the undisrupted locus, but no such instances were detected.
Discussion
Here, we have introduced IOD—a method for computationally identifying and experimentally utilizing native attB sites and their cognate integrases for genomic insertion of large DNA cargo. We have experimentally validated seven predictions in three biotechnologically relevant strains of bacteria (P. putida S12 and KT2440 and S. elongatus UTEX 2973), but IOD can be extended to a wide range of prokaryotes. These results illustrate the utility of our IOD method across phyla, and demonstrate use of both electroporation (P. putida) and conjugation methods (S. elongatus) for delivery of genetic cargo with these discovered integrases. Further, we tested integration of relatively small cargo sizes (∼6 kb in P. putida and 11 kb in S. elongatus); however, based on the size of naturally occurring GIs, we believe it likely that much larger cargos could generally be delivered using IOD-predicted integrases [12, 47].
IOD also provides access of integrase-mediated genetic editing to a diversity of nonmodel microbes. We predicted open attB sites for all but one strain when we applied our IOD software to genomic sequences from 183 strains of bacteria and archaea spanning the tree of life (182/183 tested). The IOD software can search for candidate attB sites in any genomic sequence, including eukaryotes, though successes for nonprokaryotes may be infrequent due to low sequence similarity with the prokaryote-sourced attB sites. Furthermore, utilizing integrase/att pairs that are drawn from genomes of near phylogenetic relatives to the strain of interest are less likely to require host factors or host background conditions not present in strain of interest. Lastly, while the IOD method entails using the endogenous attB sites for genome editing, the IOD database significantly increases the number of known integrase/attB pairs. This database could be mined to diversify integrases/att pairs used in other integrase-based technology such as SAGE or PASTE.
Integrases mediate stable DNA integration, but the plasticity of prokaryotic genomes may lead to loss of the inserted DNA cargo from the population over time. Our software aids in improving integration site-specificity, stability, and efficiency by filtering out attB sites present at multiple copies, those within GIs, and those whose predicted integrase appears to be defective in some way. While we performed numerous follow up experiments in P. putida to ensure our attB/integrase pairs were high quality, this step is not required and was not done for S. elongatus. However, researchers who want to further ensure that IOD integration is site-specific and stable may exclude attB sites that fall within or close to experimentally identified recombination hotspots [29]. Additionally, promiscuous native integrases present in the target strain could, if actively produced, be cross reactive with the selected IOD attP. We suggest users compare amino acid sequences to ensure that the chosen IOD integrase is distinct from native integrases in the target strain. Finally, stability can be enforced by maintaing antibiotic selection to ensure that target cell populations maintain the inserted cargo. Future software improvements may include versions flagging integrases in site-promiscuous clades and those which likely disrupt essential genes (e.g. developmental genes [5]), to further improve output candidate integrases and attB pairs for genomic integration. However, systematic studies are required to establish reliable and efficient criteria for these searches.
By utilizing attB sites native to a host, IOD bypasses the requirement for multiple transformations or insertion of attB landing pads that are needed for traditional integrase-mediated genomic integration approaches [15–17]. While integration into the endogenous attB sites may cause fitness defects, this is unlikely for a number of reasons. Many GIs restore the gene they insert into producing a functional copy that reduces any impact on bacterial physiology [5, 8]. The GIs that utilize these attB sites have persisted in bacterial genomes throughout evolutionary history [48], suggesting the fitness impact is positive or neutral for many GIs that use these sites. In most cases, synthetic integration cassettes that utilize the naturally occurring att sites predicted by IOD software can be designed to emulate the genetic neutrality of the natural GIs from which these originate. Further, as has been done with landing pad att sites, these endogenous att sites can be tested in genetically tractable organisms, established as being correct matches for the integrase, and integrase and att sites used in any organism in which the attB sites are found.
Serine integrases have traditionally been favored for biotechnological applications, due to their reduced requirements (e.g. short att sequences and no required cofactors) and greater integration stability compared to tyrosine integrases [11]. However, our test cases demonstrate that the tyrosine integrases also have great general utility, mediating genomic integration at comparable rates to the tested serine integrase. Utilizing tyrosine integrases significantly broadens the applicability of IOD, because tyrosine integrases are far more abundant in prokaryotic genomes than serine integrases (∼8-fold), which greatly expands the potential attB sites that may be present within a target organism. Furthermore, IOD enables selection of integrases from close relatives of a target strain, mitigating any potential dependencies that the integrases might have on host factors. Because of this, IOD extends the possibility of genetic engineering to virtually all prokaryotes, including those in microbiomes, to facilitate site-specific delivery of large cargo into the genome. Finally, we note that our results have been produced using att site sequences and integrase sequences exactly as found in nature, without optimization. Further work may enable increased integration efficiency, shortening and simplifying att sites, and use of att site sequence variation to increase species coverage.
Supplementary Material
gkag106_Supplemental_Files
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Parker MT, Kunjapur AM. Deployment of engineered microbes: contributions to the bioeconomy and considerations for biosecurity. Health Security. 2020;18:278–96. 10.1089/hs.2020.0010.32816583 · doi ↗ · pubmed ↗
- 2Rafeeq H, Afsheen N, Rafique S et al. Genetically engineered microorganisms for environmental remediation. Chemosphere. 2023;310:136751. 10.1016/j.chemosphere.2022.136751.36209847 · doi ↗ · pubmed ↗
- 3Kumar A, Dubey A. Rhizosphere microbiome: engineering bacterial competitiveness for enhancing crop production. J Adv Res. 2020;24:337–52. 10.1016/j.jare.2020.04.014.32461810 PMC 7240055 · doi ↗ · pubmed ↗
- 4Liu Y, Feng J, Pan H et al. Genetically engineered bacterium: principles, practices, and prospects. Front Microbiol. 2022;13:997587. 10.3389/fmicb.2022.997587.36312915 PMC 9606703 · doi ↗ · pubmed ↗
- 5Mageeney CM, Lau BY, Wagner JM et al. New candidates for regulated gene integrity revealed through precise mapping of integrative genetic elements. Nucleic Acids Res. 2020;48:4052–65. 10.1093/nar/gkaa 156.32182341 PMC 7192596 · doi ↗ · pubmed ↗
- 6Groth AC, Calos MP. Phage integrases: biology and applications. J Mol Biol. 2004;335:667–78. 10.1016/j.jmb.2003.09.082.14687564 · doi ↗ · pubmed ↗
- 7Yu SL, Mc Clain HM, Torrance EL et al. The TIGER/Islander resource for genomic islands. Microbiol Resour Announc. 2025;14, e 0049225. 10.1128/mra.00492-25.40788136 PMC 12424338 · doi ↗ · pubmed ↗
- 8Hudson CM, Lau BY, Williams KP. Islander: a database of precisely mapped genomic islands in t RNA and tm RNA genes. Nucleic Acids Res. 2015;43:D 48–53. 10.1093/nar/gku 1072.25378302 PMC 4383910 · doi ↗ · pubmed ↗