Insights into the dynamic cell associated and secreted proteome of Staphylococcus aureus cultured in TSB and milk media: a proteomic analysis
Wanting Zhu, Yujing Wang, Yanxin Li, Changjiang Zang, Hongning Jiang, Qijing Du, Jun Wang, Rongbo Fan, Rongwei Han, Yongxin Yang

TL;DR
This study compares how Staphylococcus aureus changes its proteins in different growth media, showing how nutrients affect protein expression and offering insights for food safety.
Contribution
The study reveals how culture medium affects the dynamic proteome of S. aureus, identifying potential biomarkers for food safety.
Findings
Cell-associated proteins like acetyl-CoA acyltransferase and argininosuccinate synthase increase in TSB medium over time.
Secretory proteins such as δ-hemolysin and phospholipase C are upregulated in both TSB and milk media.
Milk medium leads to increased levels of thermonuclease and SarA, while TSB medium boosts leukocidin S and staphopain B.
Abstract
The availability of nutrients is closely linked to the protein expression profile of microorganisms. In this study, a comparative investigation has been performed to examine the expression patterns of cell associated proteins and secreted proteins in Staphylococcus aureus cultivated in TSB and milk media at three time points: 3 h, 9 h, and 18 h of the growth stage using proteomic techniques. In TSB medium, cell-associated proteins such as acetyl-CoA acyltransferase, acyl-CoA synthetase, argininosuccinate lyase, and argininosuccinate synthase were detected at higher levels at 9 h and 18 h compared with 3 h, indicating overall increases during growth. While in milk, quinone oxidoreductase 1 and zinc-type alcohol dehydrogenase-like protein SAR2277 exhibited a significant increase across 3, 9 and 18 h. Secretory proteins such as δ-hemolysin, phospholipase C and panton-valentine leukocidin F…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
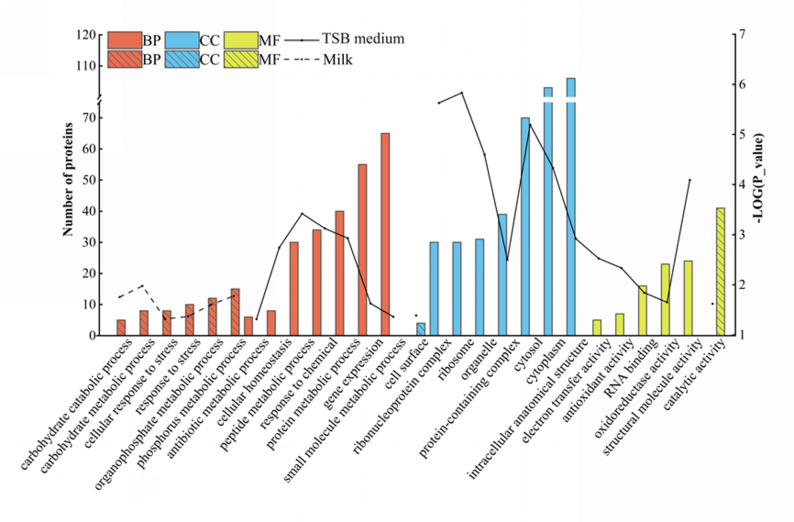 Figure 1
Figure 1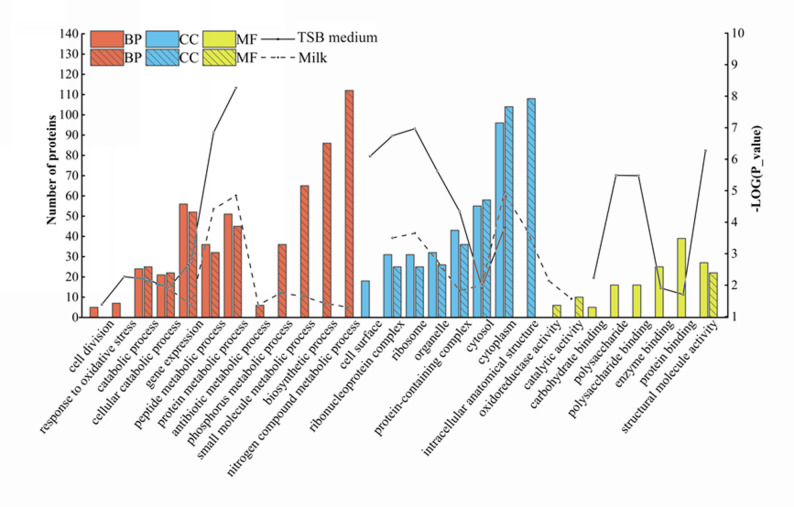 Figure 2
Figure 2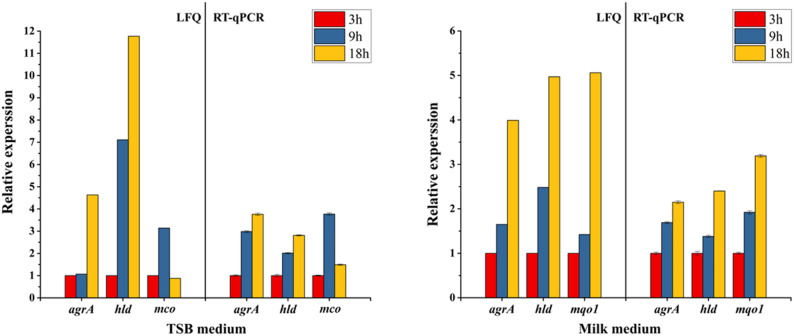 Figure 3
Figure 3- —Shandong Provincial Natural Science Foundation, China
- —https://doi.org/10.13039/501100012166National Key Research and Development Program of China
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsProbiotics and Fermented Foods · Bacterial biofilms and quorum sensing · Antimicrobial Resistance in Staphylococcus
Introduction
Staphylococcus aureus (S. aureus) is a common pathogenic bacterium frequently detected in milk [1, 2], with detection rates varying across different geographical regions and seasons [3, 4]. This bacterium plays a crucial role in altering milk composition through the enzymatic breakdown of casein, thereby impacting the quality and nutritional value of the milk [5, 6]. The ability of S. aureus to proliferate in dairy products and produce a range of harmful toxins, which pose significant health risks to consumers, has been well documented [7]. The enterotoxins produced by S. aureus exhibits significant heat resistance, such that it may survive even after high-temperature pasteurization of dairy products [8, 9]. Consequently, monitoring S. aureus levels in milk is essential for ensuring food safety and safeguarding public health.
In recent years, significant advances have been made in the research of S. aureus, encompassing its pathogenic mechanisms, drug resistance, and other related aspects [10–12]. There is also considerable interest in understanding how growth media and environmental variables influence protein expression in S. aureus. Different growth conditions have been demonstrated to influence the expression of a wide range of proteins, including those critical for metabolism, pathogenicity, and stress responses [13]. For instance, research into the effects of growth media composition on S. aureus has shown that cultivation in Dulbecco’s Modified Eagle Medium or Tryptic Soy Broth (TSB) markedly affects the bacterium’s capacity for biofilm formation [14]. At the transcriptional level, a comparison of cation-adjusted Mueller-Hinton broth and Roswell Park Memorial Institute 1640 Medium supplemented with 10% Luria broth revealed differential expression in over 800 genes [15]. At the protein level, a total of 60 differentially regulated cytoplasmic proteins were detected in S. aureus grown in TSB medium supplemented with different concentrations of NaCl, and alterations in proteins associated with protein synthesis were higher in cells grown with NaCl compared to those grown without salt [16].
The proteins of S. aureus have been categorized into several groups, including cytosolic proteins, membrane-bound proteins, cell surface-associated proteins, and extracellular proteins [17, 18]. Chronic infections, primarily associated with S. aureus extracellular proteins, persist due to the pathogen’s ability to evade host immune responses through the secretion of various virulence factors [19]. Secreted toxins, such as exotoxins, account for about 10% of the total secretome (around 1354 proteins) [20, 21] and confirmed the important role of extracellular virulence factors in the pathogenesis of S. aureus [22]. The expression of these virulence factors in S. aureus is recognized to fluctuate based on the growth environment [23]. For instance, triacylglycerol 1 and 2 were secreted by S. aureus in a NaCl-supplemented medium [16]. The capacity of S. aureus to thrive in milk is intricately linked to its pathogenicity, with enterotoxins in particular being secreted into the milk during bacterial proliferation [24, 25]. It was illustrated that although S. aureus strains produce lower levels of the staphylococcal enterotoxin D and staphylococcal enterotoxin R in milk compared to microbial broth, some strains are still capable of secreting over 2 µg/mL of staphylococcal enterotoxin D in milk [26]. As previously reported, a few studies investigated the dynamic changes in the microbial proteome and secretome of S. aureus cultured in different media [22, 27].
The aim of this study is to utilize a label-free quantitative proteomics approach to systematically investigate the differential expression of cell associated proteins and secreted proteins in S. aureus cultured in milk and TSB media at different growth stages. The results of this study may offer novel insights into the interplay between protein expression and nutritional factors, thereby contributing to the improvement of milk safety standards and quality control measures.
Materials and methods
Determination of bacterial growth curve
TSB medium (Qingdao Haibo Biotechnology Co., Ltd.), composed of 17.0 g casein tryptone, 5.0 g NaCl, 3.0 g soybean papain hydrolysate, 2.5 g di-potassium hydrogen phosphate and 2.5 g glucose, was prepared by dissolving the components in MilliQ water at a concentration of 3.0% (w/v). Skim milk powder was purchased from Inner Mongolia Yili Industrial Group, the milk powder samples were re-dissolved with MilliQ water at a ratio of 1:9 (W/V) under stirring at 40 °C for 1 h. The growth curve of S. aureus (ATCC 25923) was assessed in both TSB and milk media at 37 °C with shaking at 225 rpm/min. The milk culture was counted by plate colony counting method every hour from 0 to 10 h and every 3 h from 12 to 24 h to finalize the bacterial growth curve in milk. The growth of S. aureus in the TSB medium was monitored with 600 nm optical density values during the same period.
As illustrated in Fig. S1, the growth period of S. aureus was divided into three parts, 1–3 h for the log phase, 3–9 h for the logarithmic period, and 9–18 h for the stationary phase. Samples were taken at 3 h, 9 h, and 18 h, respectively, representing the growth of S. aureus at the transition period between phases.
Bacterial isolation
Samples were collected at 3, 9, and 18 h from S. aureus cultures grown in TSB medium. The culture was transferred to a sterile 50 mL conical centrifuge tube and centrifuged at 9,000 × g for 10 min at 4 °C to collect the bacterial cells, then washed three times with phosphate-buffered saline, followed by rapid freezing to minimize cell lysis and protein degradation. As well, the supernatant was collected for secreted proteins.
For S. aureus cultured in milk, the cultured milk was transferred to a sterile 50 mL conical centrifuge tube and centrifuged at 9,000 × g for 10 min at 4 °C to collect the bacterial cells and casein micelles, while the supernatant was retained for the extraction of secreted proteins. The cell pellet was washed 3 times with phosphate-buffered saline, then washed 3 times with 500 mM EDTA solution to dissociate from the casein micelles until the washings were clarified and centrifuged at 4 °C to retain the cells pellet. The resulting pellet was subsequently stored at −20 °C until further use.
Extraction of cell associated protein
The bacterial cells were ground in a sterile mortar with liquid nitrogen and subsequently resuspended in the SDT lysis buffer, which consists of 4% SDS (W/V), 100 mM dithiothreitol, and 100 mM Tris-HCl buffer (pH 7.6). The lysis buffer cell suspension was sonicated on ice at 200 W for 10 min (8 s on, 5 s off) using an ultrasonic cell disruptor (ATPIO XO-1000D, China). After sonication, the mixture was centrifuged at 10,000 × g for 10 min. The supernatant was collected and subjected to protein precipitation by the addition of four volumes of pre-cooled acetone, followed by incubation at −20 °C overnight. The protein pellet was obtained by centrifugation at 10,000 × g for 10 min and subsequently washed twice with pre-cooled acetone. The resulting pellet was then resuspended in 100 mM Tris-HCl buffer, and the protein concentration was determined using the Bicinchoninic Acid Assay protein assay kit (Thermo Fisher Scientific, USA).
Extraction of secreted proteins
The TSB media or milk supernatant from the isolated bacteria grown after 3 h, 9 h, and 18 h was collected respectively and mixed with an equal volume of trichloroacetic acid solution (20% w/v) to induce protein precipitation. The resulting protein precipitate was washed twice with pre-cooled acetone and subsequently reconstituted in 100 mM Tris-HCl buffer. The protein concentration was then determined as above-mentioned.
Protein digestion
Thirty micrograms of bacterial cell associated proteins and secreted proteins from each sample of species were mixed with 4% SDS and 30 mM Tris-HCl, pH 7.6, and reduced in a 100 mM dithiothreitol solution at 65 °C for 45 min. After the samples cooled, the protein solution was mixed with 200 µL UT buffer (8 M urea, 100 mM Tris-HCl, pH 8.5), applied to a 10-kDa cut-off filter tube (Sartorius, Göttingen, Germany), and centrifuged at 14,000 × g for 25 min. After washing the samples with UT buffer, alkylation was performed using 50 mM iodoacetamide in the dark at 25 °C for 45 min. The protein samples were then digested with 100 µL of trypsin solution (1 µg sequencing-grade trypsin in 50 mM ammonium bicarbonate) in a water bath at 37 °C for 16–18 h. The digestion reaction was terminated. by adding 10 µL of formic acid. Next, the peptide mixture was loaded onto a C18 column (60108–303; Thermo Fisher Scientific, MA, USA) for desalting. The samples were dried in a speed vacuum and placed at − 80 °C. Three independent biological replicates were conducted for each experimental condition.
Data-dependent acquisition analysis by LC-MS/MS
The peptide mixtures were dissolved in 0.1% formic acid and analyzed using Easy nLC 1000 chromatography coupled with an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific, CA, USA). Digested peptides were initially loaded onto a C18 trap column (100 μm × 2 cm, 5 μm; Thermo Fisher Scientific) using solution A (0.1% formic acid) at a flow rate of 300 nL/min, and subsequently separated on a C18 analytical column (100 mm × 75 μm, 3 μm; Thermo Fisher Scientific). The separation gradient was programmed with solution B (0.1% formic acid in 80% acetonitrile) as follows: 0–10% B over 3 min, 10–35% B over 45 min, 35–80% B over 26 min, ramping to 100% B within 1 min, and maintaining 100% B for an additional 15 min.
For proteomic analysis, the Orbitrap Fusion Lumos MS operated in positive ion mode with a parent ion scan range of 300–1800 m/z. Automatic switching between MS and MS/MS modes was enabled. The survey scan parameters were set to a resolution of 60,000, automatic gain control (AGC) target of 400,000, maximum injection time of 50 ms, and exclusion duration of 40 s. The top 20 most intense precursor ions with a charge ≥ 2 from the survey scan were selected for higher-energy collisional dissociation fragmentation. The MS/MS parameters included normalized collision energy of 27 eV, MS/MS resolution of 15,000, AGC target of 50,000, and maximum injection time of 50 ms.
Protein identification and quantification
The raw files were processed through MaxQuant software (version 2.0.3.0) to search against data downloaded from the UniProtKB database (S.aureus 11217 entries; downloaded December 2023) [28]. The parameters were set as follows: trypsin/P was designated as the digestion enzyme with a maximum allowance of two missed cleavage sites. Carbamidomethylation of cysteine residues was specified as a fixed modification, while N-terminal acetylation and methionine oxidation were defined as variable modifications. Matching between runs was enabled with a retention time tolerance of 0.7 min and an ion mobility tolerance of 0.05. Protein quantification was carried out using a label-free quantification (LFQ) approach, based on the intensities of razor and unique peptides. Protein and peptide identifications were conducted with a false discovery rate and peptide-spectrum match threshold set at 0.01.
Quantitative real-time PCR (RT-qPCR) analysis
The total RNA of S. aureus grown in TSB and milk at different stages was extracted from cells using a Bacteria Total RNA Isolation Kit (Sangon Biotech Co., Ltd., Shanghai, China). Total RNA was reverse transcribed and amplified using the BeyoFast™ SYBR Green One-Step qRT-PCR Kit (Beyotime, China). Four differential abundant proteins were selected and their mRNA and gyrB expression levels were determined using RT-qPCR, each gene was performed in triplicate (Table S1). Statistical analysis was conducted using SPSS (Statistical Package for the Social Sciences).
Bioinformatics and statistical analysis
Protein identification was accepted if the protein was detected in two out of three biological replicates, with each detection supported by at least two peptides, and these proteins were used for subsequent data analysis. The quantified proteins were subjected to Perseus software (www.maxquant.org/perseus/) for statistical analysis and principal component analyses (PCA). One-way analysis of variance (ANOVA) was conducted to evaluate differences in protein abundance among groups. Differentially abundant proteins were identified based on a |log_2_ (fold change) | ≥ 1 and a P-value < 0.05. These differential abundant proteins were subjected to TBtools-II (Toolbox for Biologists) v2.083 software for gene ontology enrichment and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways. The data of selected genes obtained by RT-qPCR were analyzed using one-way ANOVA followed by Duncan’s test in IBM SPSS software. P-value < 0.05 was defined as statistical significance.
Results
Identification and analysis of cell associated proteins
Based on the label free proteomics approach, a total of 14,490 peptides were identified from cell associated proteins of S. aureus cultured in TSB medium, whereas 12,259 peptides corresponding to 909 distinct proteins were detected from cell associated proteins of S. aureus grown in milk (Fig. S2). The identified cell associated proteins from both milk and TSB media were subjected to PCA. As shown in Fig. S3, the PCA score plots of cell-associated proteins in S. aureus revealed a clear separation between the two media, whereas the secreted proteins from S. aureus cultivated in milk and TSB exhibited similar expression profiles across the three growth phases (3 h, 9 h, and 18 h). These findings indicate that the composition of the culture medium exerts a pronounced influence on the overall expression patterns of cell-associated proteins in S. aureus. In this study, the selection of differentially expressed proteins was primarily based on their significant changes in abundance, with screening criteria of |log₂ (fold change) | ≥ 1 and P < 0.05.
S. aureus cell associated proteins in TSB medium
Statistical analysis of S. aureus cell associated proteins in TSB medium indicated that several proteins followed unique temporal patterns among the 3, 9, and 18 h samples. Acetyl-CoA acyltransferase (FadA), acyl-CoA synthetase (FadD), argininosuccinate lyase, argininosuccinate synthase, formate acetyltransferase, and NAD-specific glutamate dehydrogenase showed an increase in abundance across 3 h, 9 h, and 18 h. The abundance of aldehyde-alcohol dehydrogenase, N-acetylmuramoyl-L-alanine amidase sle1 (Sle1), multicopper oxidase (Mco), and pyruvate kinase increased between 3 and 9 h, then decreased between 9 and 18 h growth. Moreover, the abundances of translation initiation factor IF-1, adapter protein MecA, tetratricopeptide repeat protein, 30 S ribosomal protein S18, and fructose-1,6-bisphosphate aldolase decreased between 3 and 18 h.
S. aureus cell associated proteins in milk medium
In milk medium, the abundance of several proteins, including quinone oxidoreductase 1 (Mqo1), carbamate kinase 1, and zinc-type alcohol dehydrogenase-like protein SAR2277 exhibited a significant and consistent increase from 3 to 18 h. The abundance of transketolase increased between 3 and 9 h but decreased between 9 and 18 h. Conversely, the abundance of DHHA1 domain protein and proline/betaine transporter decreased between 3 and 9 h and then increased between 9 and 18 h. Meanwhile, the protein abundance of cold shock protein CspA, cell-division initiation protein and delta-hemolysin decreased between 3 and 18 h.
Comparative analysis of S. aureus cell associated proteins in TSB and milk media
Regarding the differences in S. aureus cell associated proteins between media, the levels of thiol peroxidase, 4-hydroxy-tetrahydrodipicolinate synthase, and tyrosine–tRNA ligase were higher in TSB medium than in milk between 9 and 18 h, suggesting more active cellular metabolism under nutrient-rich TSB conditions. However, several proteins, such as acyl carrier protein, Sle1, and phosphatidylglycerol–prolipoprotein diacylglyceryl transferase were greater in milk than in TSB medium between 9 and 18 h, indicating that S. aureus adapts to the milk environment by enhancing its cell envelope integrity and modifying its surface structures to cope with environmental stress and nutrient limitation.
Protein annotations were used to classify differentially abundant S. aureus cell associated proteins into biological processes, cellular components, and molecular functions. At the biological process level, the most abundant proteins in TSB medium were primarily associated with response to chemical, cellular homeostasis, small molecule metabolic process, protein metabolic process, and gene expression. In contrast, in milk, the predominant processes included response to stress, phosphorus metabolic process, carbohydrate metabolic process, organophosphate metabolic process, and carbohydrate catabolic process. At the cellular component level, in TSB, the majority of proteins were associated with the protein-containing complex, ribonucleoprotein complex, cytosol, organelle, cytoplasm, intracellular anatomical structure, and ribosome, whereas in milk, proteins were primarily involved in the cell surface. Regarding molecular functions, in TSB, the most common functions were structural molecule activity, electron transfer activity, antioxidant activity, RNA binding, and oxidoreductase activity, while in milk, catalytic activity was more prominent (Fig. 1).Fig. 1. Gene ontology analysis of differentially abundant cell associated proteins of S. aureus cultured in TSB and milk media. The solid and dashed lines represent − log(P-value) of functional enrichment significance in TSB medium and milk, respectively. BP, biological process; CC, cellular component; MF, molecular function
The KEGG pathway analysis of differentially abundant proteins in S. aureus cultivated in TSB medium and milk is summarized in Table S2. The differential abundance of cell associated proteins under these two growth conditions was associated with a variety of pathways, including carbohydrate metabolism, translation, energy metabolism, pyruvate metabolism, glycolysis/gluconeogenesis, chaperones and folding catalysts. Notably, specific pathways such as amino acid metabolism, nucleotide metabolism, purine metabolism, the citric acid cycle, and xenobiotics biodegradation and metabolism were predominantly identified in the TSB medium. In contrast, the milk medium showed strong associations with pathways including transfer RNA biogenesis, exosome, mitochondrial biogenesis, aminoacyl-tRNA biosynthesis, oxidative phosphorylation and carbon fixation in photosynthetic organisms.
Identification and analysis of secreted proteins
Secreted proteins of S. aureus in TSB medium
In TSB medium, a total of 6,985 peptides corresponding to 552 secreted S. aureus proteins were identified, whereas 4,605 peptides corresponding to 480 secreted proteins were detected during bacterial growth in milk (Fig. S4). We quantified secreted proteins from both milk and TSB media. These proteins were subjected to PCA analysis. The PCA score plots showed distinct clusters for each of the three stages: 3 h, 9 h, and 18 h. This clustering occurred in both media. The results indicated a clear separation of protein profiles associated with proteins released at different stages in TSB and milk media (Fig. S3).
The relative abundances of these proteins varied significantly over time. In TSB medium, peptidase T (PepT), staphopain B, phospholipase C (Hlb), triacylglycerol lipase, γ-hemolysin component B (HlgB), delta-hemolysin (Hld), bifunctional autolysin, and panton-Valentine leukocidin F (LukF-PV) demonstrated an increase in abundance from 3 to 18 h. Moreover, the levels of superoxide dismutase [Mn/Fe] (SodM), type VII secretion system extracellular protein A (EsxA), and clumping factor A increased significantly between 3 and 9 h, then decreased between 9 and 18 h. The protein abundance of bacilliredoxin SAS1371, UDP-N-acetylglucosamine 1-carboxyvinyltransferase 1, iron-sulfur cluster carrier protein, and uridylate kinase decreased between 3 and 18 h.
Secreted proteins of S. aureus in milk medium
In the milk medium, the secreted proteins of phospholipase C, Hlb, HlgB, Hld, transcriptional regulator SarA (SarA), glyoxal reductase, and LukF-PV increased abundance in milk across 3, 9, and 18 h. The abundance of thermonuclease (Nuc) was observed to increase significantly between 3 and 9 h, and then remain unchanged between 9 and 18 h. In addition, the abundance of ornithine carbamoyltransferase, iron-sulfur cluster repair protein ScdA, and EsxA increased between 3 and 9 h and decreased between 9 and 18 h. Furthermore, the abundance of SodM, 30 S ribosomal protein S19, and iron-sulfur cluster carrier protein decreased between 3 and 18 h.
Comparative analysis of secreted proteins of S. aureus in TSB and milk media
Throughout the growth stages, the abundance of several proteins, including urease subunit alpha (UreC), Hlb, LukF-PV, HlgB, and Hld, increased significantly across 3, 9, and 18 h in both TSB and milk media. In addition, the levels of PepT, staphopain A, and cell division protein SepF were greater in TSB medium than in milk at 9 h and 18 h, while several proteins, such as oligoendopeptidase F, SarA, and serine-aspartate repeat-containing protein D were higher in milk than in TSB medium.
Protein annotations revealed that the differentially abundant secreted proteins of S. aureus in both TSB and milk were mainly associated with the functions such as metabolic and catabolic process, cytosol and cytoplasm components, structural molecule activity (Fig. 2).Fig. 2. Gene ontology analysis of differentially abundant secreted proteins of S. aureus cultured in TSB and milk media. The solid and dashed lines represent the − log(P-value) of functional enrichment significance in TSB medium and milk, respectively. BP, biological processes; CC, cellular components; MF, molecular functions
Table S3 summarizes the KEGG pathway analysis of differentially abundant secreted proteins of S. aureus in TSB and milk media. Overall, these proteins were associated with carbohydrate metabolism, translation, amino acid metabolism, energy metabolism, ribosome, pyruvate metabolism, exosome, membrane trafficking, glyoxylate and dicarboxylate metabolism pathways. Notably, several pathways were uniquely enriched in TSB, including pyrimidine metabolism, bacterial toxins, translation factors, lipid biosynthesis, nicotinate and nicotinamide metabolism, RNA polymerase and transcription. In contrast, pathways related to lipid metabolism, xenobiotic biodegradation and metabolism, nucleotide metabolism, the pentose phosphate pathway, and benzoate degradation were predominantly enriched in the milk medium.
Supporting the proteomic data at the mRNA level
To support and complement the proteome data obtained through LFQ analysis, the expression patterns of selected proteins and their corresponding genes were analyzed using quantitative reverse transcription polymerase chain reaction (RT-qPCR). The results showed a time-dependent increase in the expression of the AgrA and Hld genes in TSB medium across 3, 9 and 18 h. Furthermore, mco expression increased between 3 and 9 h and subsequently declined between 9 and 18 h in the TSB medium. In contrast, ureC, hld, and mqo1 expression rose consistently in the milk medium over time (Fig. 3). These expression profiles were relatively consistent with the LFQ proteomic results, showing a link between protein abundance variations and gene expression levels.Fig. 3RT-qPCR analysis of genes corresponding to proteins identified in the proteomic analysis of S. aureus grown in either milk or TSB media
Discussion
Changes in cell associated proteins of S. aureus both conditions
The protein abundances of FadA and FadD increased in TSB medium across 3, 9 and 18 h. Consistent with previous studies using microarray analysis, the FadA and FadD genes were highly elevated within two days of cultivation [29]. Specifically, FadA was responsible for conjugating long-chain fatty acids with coenzyme A to produce fatty acyl-CoA, which played a critical role in lipid synthesis and repair by facilitating the transport of fatty acyl groups [30]. FadD, on the other hand, catalyzes the conversion of free fatty acids into fatty acyl-CoA by binding with Coenzyme A (CoA), a process known as fatty acid “activation”, which is the first step in fatty acid metabolism. Fatty acyl-CoA is the active form of fatty acids, enabling their further metabolism through various pathways, such as β-oxidation (which breaks down fatty acids into acetyl-CoA) and fatty acid synthesis [31]. Moreover, the increased expression of FadA and FadD indicated that S. aureus was actively engaging in fatty acid metabolism to adapt to the nutritional circumstances in TSB medium, perhaps boosting its adaptability.
In TSB medium, the abundance of Mco was significantly higher at 9 h compared to 3 h, followed by a notable decrease at 18 h. Previous research used TSB medium to culture S. aureus and quantitatively analyze Mco transcript levels using RT-qPCR, indicating a considerable rise in Mco expression during the growth phase [32]. The elevated Mco expression during the pre-growth phase could be associated with oxidative stress [33]. Mco, an important antioxidant enzyme in bacteria, is involved in the oxidative stress pathway, where redox equilibrium within cells is effectively maintained through the catalysis of superoxide dismutation and the regulation of the oxidation states of metal ions, thereby mitigating their toxicity [34]. In addition, Mco is involved in the copper homeostasis pathway, where bacteria acclimate to growth conditions by maintaining internal copper levels at the lowest concentration required for growth, which results in a decrease in abundance of Mco proteins [35].
The abundance of Mqo1 increased significantly in milk across 3, 9 and 18 h. Quantitative qRT-PCR analysis demonstrated a marked upregulation of mqo1 expression over a 24-hour period in TSB supplemented with 0.5% glucose [36]. Mqo1, an enzyme engaged in the TCA cycle, was responsible for oxidizing malate to oxaloacetate, which is necessary for gluconeogenesis [37]. Notably, the chemical composition of TSB medium contains a higher concentration of glucose compared to milk. The significant increase of Mqo1 expression in milk may be attributable to the lack of immediately available glucose for S. aureus in milk, requiring the bacterium to metabolize alternative substrates to create energy for growth and reproduction. The cell-associated differential protein such as Mqo1 may serve as potential biomarkers for evaluating milk quality and early monitoring of S. aureus infected with dairy cows.
Changes in secreted proteins of S. aureus under different growth conditions
Both TSB and milk are nutritionally rich environments that support the vigorous growth of S. aureus. Accordingly, secreted proteins associated with biological processes and cellular components were markedly upregulated in both media, reflecting the metabolically active state of S. aureus under nutrient-abundant conditions. The abundance of UreC showed a significant increase across 3, 9 and 18 h in both milk and TSB media. Furthermore, UreC expression in S. aureus was dramatically enhanced during the exponential growth phase when cultivated in Chemically Defined Medium, as shown by proteomic studies [38]. UreC is an essential enzyme in the urea metabolism pathway, catalyzing the hydrolysis of urea to yield ammonia and carbon dioxide. This process helps to regulate the pH within cells and promote energy metabolism through acetyl-CoA synthetase and the tricarboxylic acid cycle, providing the essential energy for cell development and adaptability [39]. The enzymatic activity of UreC was critical for maintaining the energy balance needed for cell development and adaptability.
A significant increase in the abundance of hemolysin-related Hld, Hlb and HlgB was observed between 3 and 18 h in both milk and TSB media, aligning with a high expression of Hld mRNA in S. aureus during both the growth and stationary phases cultured in LB medium [40]. Most S. aureus isolates produce hemolysin, which is generally recognized as a key element in the pathogenicity of infections caused by these bacteria [41, 42]. Hemolysin was recognized to be selectively interacting with the phospholipid bilayer of host cell membranes, being introduced into the membrane and producing modifications in its structure and permeability. This process causes the creation of ion channels or pores, which facilitates the passage of ions and small molecules, disturbing the internal and exterior cellular environments and eventually leading to cell lysis [43, 44]. These disturbances played an important role in the infection process, facilitating bacterial entry and survival within the host. The abundance of LukF-PV was significantly elevated in both milk and TSB media across 3, 9 and 18 h; however, its expression levels did not differ significantly between the two conditions at corresponding time point. In a previous study, LukF-PV was considerably elevated during the growth phase of S. aureus cultured in BHI medium using quantitative RT-PCR [45]. LukF-PV has been shown to bind specifically to host leukocytes, where it facilitates the formation of transmembrane pores or channels in the cell membrane. The resulting disruption of ionic and molecular homeostasis across the membrane ultimately leads to leukolysis [46]. Furthermore, LukF-PV was found to induce macrophage death, mediate the escape of S. aureus from the host cell, thereby facilitating the survival of S. aureus [47]. Thus, an increased abundance of LukF-PV augments S. aureus virulence by targeting and lysing host immune cells, thereby facilitating bacterial survival and persistence. Interestingly, the expression of LukF-PV is regulated by several transcriptional regulators, including two-component systems, quorum sensing mechanisms, and the accessory gene regulator (Agr) system [48]. In the present study, despite the elevated expression of Agr in milk, comparable levels of LukF-PV were observed in both milk and TSB media. The similar expression of LukF-PV across these two distinct environments suggests that the bacterial regulatory mechanisms governing toxin production in S. aureus are resilient and effectively operational in diverse conditions. This finding suggests that LukF-PV production in S. aureus is governed by multifactorial regulatory mechanisms that enable toxin synthesis under diverse environmental conditions, underscoring its potential role in the pathogenicity of S. aureus. However, further investigations are required to elucidate the precise regulatory mechanisms involved.
Nevertheless, the compositional disparities between TSB and milk created distinct metabolic interactions between the microbes and their environment. SodM secretion in TSB medium increased significantly between 3 and 9 h, then decreased between 9 and 18 h, whereas it decreased in milk across 3, 9 and 18 h. The RT-qPCR study revealed a considerable increase in SodM expression over 24 h of S. aureus growth in TSB [49]. S. aureus contains two superoxide dismutases, SodA and SodM, with SodM playing an important role in mitigating the detrimental effects of toxic superoxide anion radicals produced within the bacteria [50]. SodM, an enzyme distinct to S. aureus, catalyzes the dissociation of superoxide anion radicals into molecular oxygen and hydrogen peroxide [51], thereby reduces oxidative stress damage to bacteria, maintains intracellular redox balance, and preserves their normal physiological state [52]. Although these radicals are produced within the bacterial cytoplasm, their generation and the associated oxidative stress can be modulated by the extracellular environment. The decline of SodM of S. aureus in milk could be explained by the low concentration of superoxide anion radicals present in this medium. Antioxidant components found in milk, such as vitamin C and E compounds, successfully scavenged these free radicals, eliminating the need for SodM activity in this environment [53]. Additionally, PepT is more abundant in TSB than in milk medium, where it primarily catalyzes the removal of the N-terminal amino acid from most tripeptides. PepT1 and PepT2 have been shown to be non-essential for nutrient acquisition in peptide-rich media; however, their expression is regulated by the global virulence factor regulator SarA, implicating them as key contributors to the virulence of several bacterial pathogens [54]. Furthermore, pathways associated with bacterial toxins, human diseases, and S. aureus infection were also enriched in TSB, indicated by active protein synthesis and metabolic processes that promote the production of virulence factors. These further support the enhanced expression of virulence factors, such as hemolysins and lipases, which are pivotal in the pathogenicity of S. aureus [55]. These findings collectively underscore the dynamic nature of S. aureus metabolism, emphasizing how nutrient-rich environments like TSB enhance its pathogenic potential. The analysis provides valuable insights into how S. aureus adapts to its surroundings, optimizing the expression of virulence factors that are critical for its ability to cause infections.
The relative abundances of Nuc increased significantly between 3 and 9 h, then it remained unchanged between 9 and 18 h in milk. S. aureus cultivated in BHI broth showed an observable increase of Nuc expression during the mid-logarithmic growth phase, which was confirmed by quantitative RT-PCR [56]. The staphylococcal nuclease, which is controlled by the Nuc gene, acted as a thermostable nucleic acid enzyme capable of hydrolyzing DNA and RNA within host cells, hence contributing to tissue damage and increasing the pathogenicity of S. aureus by facilitating its spread and colonization [57]. Moreover, the ability of the nuclease to break down host nucleic acids served as a crucial nutrient source for S. aureus, with the released nucleotides and nucleic acids from host tissues being ingested by the bacterium to maintain their growth and survival within the host environment [58]. Specific growth conditions altered the regulation of Nuc expression, with the gene being upregulated during distinct growth phases, particularly in environments with limited nutrients or under conditions that mirrored the host’s physiological state [59]. Conversely, in the nutrient-rich environment of TSB, Nuc expression may have been repressed because the bacteria could receive sufficient nutrients without breaking down host nucleic acids. This suggests that the regulation of Nuc expression is linked to the availability of external resources, with nutrient-rich conditions reducing the necessity for the bacterium to engage in host tissue degradation for nutritional acquisition. Additionally, the enrichment of pathways related to lipid metabolism, xenobiotic biodegradation, and nucleotide metabolism indicates metabolic reprogramming that enables S. aureus to efficiently utilize milk-derived substrates such as fatty acids and nucleotides. However, the molecular mechanisms underlying the ability of S. aureus to adapt to nutrient availability in the milk environment and to modulate protein biosynthesis, thereby promoting its persistence and pathogenic potential, require further investigation using targeted and integrative omics approaches. The secreted proteins such as Hld, LukF-PV, and Nuc, which are bacterial toxins produced by S. aureus, deserve particular attention in the dairy industry, as they could help assess the hygienic status and S. aureus infection. Such biomarkers may provide valuable targets for early detection and management strategies aimed at preventing contamination and controlling the infection of S. aureus, as well as for evaluating the risk of toxin-associated foodborne illnesses. Nevertheless, further experimental validation is required to assess the potential health risks of these proteins to consumers.
The observed proteomic alterations likely represent the outcome of complex interactions between S. aureus and its surrounding culture environment. The increased expression of acyl-CoA synthetase, Mco, and staphopain A in TSB, along with the elevated abundance of Mqo1 and Nuc in milk, reflects metabolic reprogramming that enhances energy production and oxidative stress responses to support bacterial survival. These proteomic shifts align with the enrichment of pathways related to lipid biosynthesis, energy metabolism, and nucleotide metabolism, indicating that S. aureus flexibly modulates its metabolic network to utilize available nutrients of each medium. Collectively, the divergent proteomic profiles observed in milk and TSB represent environment-specific adaptive responses to nutrient composition and metabolic demands. This interactive regulation highlights that the proteomic landscape of S. aureus in milk results from a dynamic bacterium–environment interplay, underscoring the pivotal role of environmental context in shaping bacterial metabolism, physiology, and pathogenic potential.
Conclusion
Dynamic changes in cell associated and secreted proteins of S. aureus cultured in both TSB and milk media were investigated using a proteomic approach. The results demonstrated that several cell associated proteins and secretory proteins, such as Hld, Hlb, and LukF-PV were increased in TSB and milk media across 3, 9 and 18 h. Notably, several virulence factors, including SarA and Sle1, were found to be present at higher levels in milk than in TSB medium, while Nuc was uniquely identified in milk. This highlighting the potential risk posed by S. aureus in milk and indicating its possible impact on food safety and consumer health. These results suggest that the nutrient composition of milk drives distinct metabolic and functional adaptations in S. aureus, promoting the preferential expression of stress response pathways and virulence factors. This study provides important insights into the metabolic reprogramming and functional adaptation of S. aureus under different growth conditions. Given that a specific isolate was used, future studies will expand the analytical framework to include a broader range of serotypes and antimicrobial resistance profiles, analyzed through proteomics, to obtain more robust and generalizable results. Overall, these findings enhance our understanding of the dynamic changes in the S. aureus secretome and have important implications for future preventive and control strategies, thereby enriching our understanding of the evolving cell associated protein and secreted protein profiles of S. aureus.
Supplementary Information
Supplementary Material 1.
Supplementary Material 2.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Foster TJ. Surface proteins of Staphylococcus aureus. Microbiol Spectr. 2019;7(4). 10.1128/microbiolspec.gpp 3-0046-2018.10.1128/microbiolspec.gpp 3-0046-2018 PMC 1095722131267926 · doi ↗ · pubmed ↗
- 2Tam K, Torres VJ. Staphylococcus aureus secreted toxins and extracellular enzymes. Microbiol Spectr. 2019;7(2). 10.1128/microbiolspec.gpp 1123-0039-2018.10.1128/microbiolspec.gpp 3-0039-2018 PMC 642205230873936 · doi ↗ · pubmed ↗
- 3Alreshidi M, Dunstan M, Macdonald RHM, Singh MK, Roberts VK. Analysis of cytoplasmic and secreted proteins of Staphylococcus aureus revealed adaptive metabolic homeostasis in response to changes in the environmental conditions representative of the human wound site. Microorganisms. 2020;8(7). 10.3390/microorganisms 8071082.10.3390/microorganisms 8071082 PMC 740916232698515 · doi ↗ · pubmed ↗
- 4Zheng X. The role of the cell envelope in Staphylococcus aureus protein secretion and resistance to host antimicrobial proteins. New York University; 2022.