Malaria-GENOMAP: a web-based tool for exploring genomic variation of malaria parasites
Joseph Thorpe, Nina Billows, Gabrielle C Ngwana-Joseph, Amy Ibrahim, Deborah Nolder, Colin J Sutherland, Thi Hong Ngoc Nguyen, Thi Huong Binh Nguyen, Quang Thieu Nguyen, Jamille G Dombrowski, Silvia Maria Di Santi, Claudio R F Marinho, Jody E Phelan, Tomasz Kurowski

TL;DR
Malaria-GENOMAP is a web tool that helps researchers explore genomic data from various malaria parasites to better understand their biology and drug resistance.
Contribution
The novel contribution is a web-based platform integrating genomic and geographic data for multiple Plasmodium species.
Findings
Malaria-GENOMAP integrates genomic variant data from six Plasmodium species.
The tool enables exploration of population structure and drug resistance markers.
It supports geographic and gene-level analysis of mutations.
Abstract
Malaria, caused by Plasmodium parasites, imposes a significant public health burden. While Plasmodium falciparum remains the primary target of elimination strategies due to its high mortality rate, lesser-known species such as P. malariae, P. vivax, and P. knowlesi continue to contribute to substantial human morbidity. Genomic approaches, including whole-genome sequencing, offer powerful tools for understanding the biology, transmission, and emerging drug resistance of these neglected Plasmodium species. However, there is an urgent need for informatic tools to summarize and visualize the high-dimensional and complex genomic data generated. We developed Malaria-GENOMAP, a user-friendly web-based tool, which integrates genomic variant data, such as allele frequencies, with geographical maps and chromosome-wide to gene views for in-depth exploration. The tool includes variation from P.…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
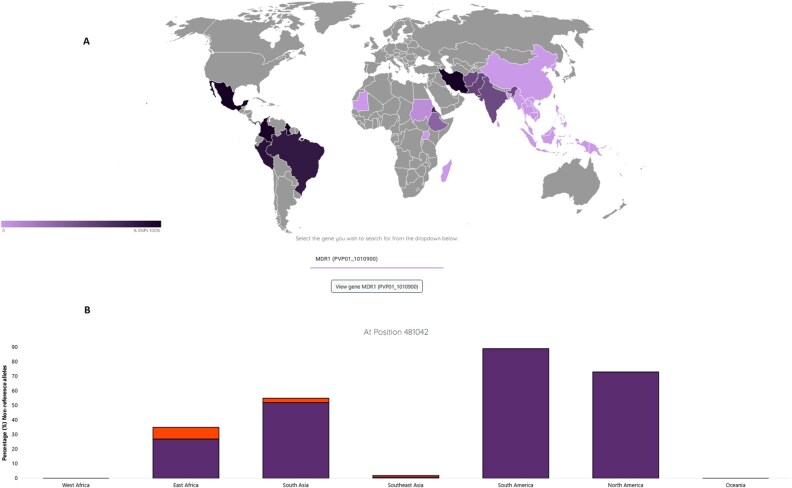 Figure 1
Figure 1| Species | Genome size | No. SNPs | No. isolates | No. countries |
|---|---|---|---|---|
|
| 24.4 Mbp | 2 051 729 | 158 | 2 |
|
| 29.6 Mbp | 221 656 | 139 | 26 |
|
| 36.0 Mbp | 268 850 | 36 | 16 |
|
| 34.3 Mbp | 216 320 | 47 | 22 |
|
| 21.7 Mbp | 123 902 | 38 | 1 |
|
| 24.2 Mbp | 535 146 | 1359 | 29 |
- —BBSRC LIDo PhD studentship
- —UKRI MRC
- —EPSRC10.13039/501100000266
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMalaria Research and Control · vaccines and immunoinformatics approaches · Calpain Protease Function and Regulation
1 Introduction
Malaria, caused by Plasmodium parasites, led to 282 million cases and 610 000 deaths across 85 endemic countries in 2024 (World Health Organization 2025). While P. falciparum accounts for most global malaria mortality, neglected species like P. vivax, P. simium, P. knowlesi, P. ovale curtisi, P. ovale wallikeri, and P. malariae also cause significant disease. Among these, P. vivax is the most widespread outside Africa, causing over 14 million cases annually (World Health Organization 2025), while P. knowlesi is found in primates throughout Southeast Asia, and is the most common cause of human malaria in Malaysia (Singh et al. 2004, Turkiewicz et al. 2023). Diagnostic challenges often underestimate the prevalence of P. malariae and P. ovale spp parasites, which are commonly found in co-infections with P. falciparum (Tajebe et al. 2014). Furthermore, neglected malaria species contribute to elimination challenges through diverse biological traits, including chronic infections in P. malariae, hypnozoite-mediated relapses in P. ovale spp, zoonotic transmission, and the presence of asymptomatic carriers (Higgins et al. 2024).
Research on these species is limited, in part due to a lack of in vitro culture systems, but whole-genome sequencing (WGS) has advanced the understanding of parasite biology and epidemiology (Phelan et al. 2023). WGS technologies have expanded insights into genome diversity, drug resistance, and population structure for P. falciparum and neglected species. Tools like selective whole-genome amplification and molecular barcodes have enabled sequencing from low-parasitaemia samples and supported surveillance efforts (Benavente et al. 2021, Ibrahim et al. 2023, Ibrahim et al. 2024, Turkiewicz et al. 2023, Ngwana-Joseph et al. 2024). However, a gap remains in tools for analyzing and summarizing genomic data for neglected malaria species, as existing platforms focus predominantly on P. falciparum and typically require substantial bioinformatics expertise to explore genomic variation across geographic regions (e.g. https://apps.malariagen.net/apps/pf7/). Other resources that include genomic data from neglected Plasmodium species, such as PlasmoDB (https://plasmodb.org/plasmo/app), contain more limited population-level data and are primarily oriented towards genome annotation rather than population structure, and are largely targeted at malaria biology specialists. Overall, there is a lack of interactive, geography-aware tools that enable non-specialist users to explore population structure, allele frequencies, and putative drug resistance markers across the various neglected Plasmodium species.
To address this, we present Malaria-GENOMAP, a web-based tool aggregating data from >1700 Plasmodium genomes, including SNPs in drug resistance genes and allele frequencies across endemic regions. Designed for neglected species, the tool supports diagnostics, drug development, and surveillance activities.
2 Methods
2.1 Malaria-GENOMAP database
The dataset includes clinical isolates of Plasmodium from single-species infections, encompassing: P. malariae (n = 158) (Ibrahim et al. 2024), P. vivax (n = 1359) (Ibrahim et al. 2023, Ngwana-Joseph et al. 2024), P. simium (n = 38) (Manko et al. 2025), P. ovale spp (wallikeri n = 47; curtisi n = 36) (Higgins et al. 2024), and P. knowlesi (n = 139) (Turkiewicz et al. 2023) (Table 1). To identify SNPs and insertions or deletions (indels), raw FASTQ files were mapped to reference genomes using bwa-mem software (v0.7.12) (Li and Durbin 2009). Variants were called with GATK’s HaplotypeCaller (v4.1.4.1) (McKenna et al. 2010), using the -ERC GVCF option to generate a combined VCF file for all isolates. This file was filtered to include only SNPs in core genomes and further refined by excluding SNPs with a negative Variant Quality Score Log-Odds. Where training SNP datasets were unavailable, SNPs were filtered using the GATK VariantFiltration function. Annotated species-specific VCF files were indexed with existing GFF3 annotation data. Associated metadata, such as collection year and location, were compiled into a TSV file. Finally, SNP and metadata were merged into a MySQL database for each species, enabling efficient user-driven queries (Fig. 1, available as supplementary data at Bioinformatics online).
Malaria-GENOMAP Screenshots. (A) Countries represented with P. vivax data; (B) Pvmdr1 gene (position 481042 T -> C; C698S) shows population differentiation between South America and Southeast Asia P. vivax populations (%).
2.2 Malaria-GENOMAP framework
The database is visualized through an intuitive interface built with the Angular framework and amCharts (2024) (https://www.amcharts.com/javascript-charts). Backend operations, powered by Node.js, retrieve and display JSON-formatted data from the MySQL database with minimal query response times. Tables are indexed for rapid searches, and users can interact with three main views:
Country: Displays data availability and annotated genomic variations (e.g. amino acid changes) for specific countries.Gene: Allows searches by locus or variant, showing global frequencies of occurrence by country, with outputs in tabular format.Alignment: Integrates IGV tools to examine variants within genomic regions, providing quality metrics and expandable annotation tracks.
The platform is accessible via https://genomics.lshtm.ac.uk/malaria-genomaps/#/, offering dynamic maps, graphs, and tables to support diverse research and surveillance needs (Fig. 2, available as supplementary data at Bioinformatics online).
3 Results
To highlight the functionality of Malaria-GENOMAP, we show some examples of its use. Markers of population differentiation may be useful for molecular barcoding and can be driven by drug resistance or mosquito vector diversity. For P. vivax, previous work has found selective sweeps proximal to pvmdr1, a putative marker for chloroquine resistance (Ngwana-Joseph et al. 2024). Using Malaria-GENOMAP, we highlight a nonsynonymous SNP leading to amino acid substitution 698S > 698G in pvmdr1 (445/1294, 34.4%), which is near fixed in South American population (264/297, 88.9%) compared to Southeast Asia (7/550, 1.3%) (Fig. 1), consistent with previous studies (Ibrahim et al. 2023). Similarly, the PVP01_1313400 K841N mutation is highly frequent in East Africa (Ethiopia 135/137, 98.5%; Eritrea 11/13, 84.6%; Uganda 3/5, 60%), but absent in Asia and South America (0/297, 0%) (Fig. 3, available as supplementary data at Bioinformatics online) (Benavente et al. 2021). Whilst the PvP47 and PvP48/45 genes are linked to the mosquito vector, with P47 K27E mutation (P48/45 R418K) found near fixed in South America (278/297, 93.6%) but absent elsewhere (0/1062, 0%) (Fig. 3, available as supplementary data at Bioinformatics online) (Benavente et al. 2021).
The pfdhfr gene in P. falciparum has been linked to pyrimethamine resistance (Turkiewicz et al. 2020), with five amino acid variants within the pmdhfr (A15S, S49R, F57L, R58S and N114S) in P. malariae aligning closely with mutations linked to drug susceptibility in pfdhfr (Ibrahim et al. 2024). The pmdhfr N114S mutation is prevalent globally (114/151, 75.5%), with high frequencies in mid-Africa (27/33, 81.8%), West Africa (23/29, 79.3%), and South America (6/6, 100%), with high-quality allele calls checked using the IGV view (Fig. 4, available as supplementary data at Bioinformatics online). This activity demonstrates the potential for Malaria-GENOMAP to discover any future SNP mutations in neighbouring regions, as well as identify new SNPs that can be linked to drug resistance.
4 Discussion
Malaria remains a critical public health issue, with genomics research largely concentrated on P. falciparum, the deadliest of the Plasmodium species, evidenced by existing tools that focus primarily on this species [https://apps.malariagen.net/apps/pf7/; Pf-HaploAtlas (Lee et al. 2024)]. This focus often leaves other Plasmodium species underrepresented, which presents challenges for achieving WHO’s malaria elimination targets (World Health Organization 2025). With advancements in sequencing technology now enabling data generation from infections with low parasitaemia, particularly for neglected malaria species, our work aims to fill this gap by providing and visualizing comprehensive genomic data. Our web-based tool, Malaria-GENOMAP, allows non-specialist users to interactively explore population structure, allele frequencies, and putative drug-resistance markers across multiple neglected Plasmodium species. These insights shed light on genetic variations that are essential for understanding infection control and population diversity (Phelan et al. 2023, Ibrahim et al. 2024). The tool aggregates and visualizes SNP data from >1700 high-quality samples, highlighting mutations linked to drug resistance and their regional specificity. This information supports the development of molecular barcodes, which can track transmission patterns, and informs the design of improved diagnostics and vaccines (Preston et al. 2014, Benavente et al. 2020). Looking forward, Malaria-GENOMAP is designed to incorporate additional data from neglected malaria species, with the potential to expand to include new functionalities such as predictive modelling for drug resistance mutations. For instance, advanced machine learning algorithms could be integrated to anticipate resistance patterns before they emerge, informing more targeted interventions.
In summary, Malaria-GENOMAP is positioned to play a role in advancing malaria research by illuminating genomic variation across neglected Plasmodium species. This tool provides invaluable resources for research and surveillance efforts focused on eradicating malaria and reducing its global health burden.
Supplementary Material
btag016_Supplementary_Data
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1am Charts: Java Script Charts. am Charts. Accessed 10 December 2024.
- 2Benavente ED , Campos M, Phelan J et al A molecular barcode to inform the geographical origin and transmission dynamics of plasmodium vivax malaria. P Lo S Genet 2020;16:e 1008576.10.1371/journal.pgen.100857632053607 PMC 7043780 · doi ↗ · pubmed ↗
- 3Benavente ED , Manko E, Phelan J et al Distinctive genetic structure and selection patterns in Plasmodium vivax from South Asia and East Africa. Nat Commun 2021;12:3160.34039976 10.1038/s 41467-021-23422-3PMC 8154914 · doi ↗ · pubmed ↗
- 4Higgins M , Manko E, Ward D et al New reference genomes to distinguish the sympatric malaria parasites, Plasmodium ovale curtisi and Plasmodium ovale wallikeri. Sci Rep 2024;14:3843.38360879 10.1038/s 41598-024-54382-5PMC 10869833 · doi ↗ · pubmed ↗
- 5Ibrahim A , Mohring F, Manko E et al Whole genome sequencing of Plasmodium malariae identifies continental segregation and mutations associated with reduced pyrimethamine susceptibility. Nat Commun 2024;15:10779.39738025 10.1038/s 41467-024-55102-3PMC 11685946 · doi ↗ · pubmed ↗
- 6Ibrahim A , Manko E, Dombrowski JG et al Population-based genomic study of Plasmodium vivax malaria in seven Brazilian states and across South America. Lancet Reg Health Am 2023;18:100420.36844008 10.1016/j.lana.2022.100420 PMC 9950661 · doi ↗ · pubmed ↗
- 7Lee C , ÜnlüES, White NFD et al Pf-Haplo Atlas: an interactive web app for spatiotemporal analysis of Plasmodium falciparum genes. Bioinformatics 2024;40:btae 673.39565917 10.1093/bioinformatics/btae 673PMC 11588202 · doi ↗ · pubmed ↗
- 8Li H , Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009;25:1754–60.19451168 10.1093/bioinformatics/btp 324PMC 2705234 · doi ↗ · pubmed ↗