CYP2C19 genotyping and mavacamten: predicting outcomes in normal, intermediate and rapid metabolisers in obstructive hypertrophic cardiomyopathy
Yande Kasolo, Edward Burford, Mohammed Obeidat, Glenda M Beaman, Thomas Monk, Rachel Bastiaenen, William G Newman, Robert M Cooper

TL;DR
This study examines how different CYP2C19 gene types affect outcomes in patients with obstructive hypertrophic cardiomyopathy taking the drug mavacamten.
Contribution
The study reveals that CYP2C19 gene variations do not significantly influence clinical outcomes in patients on mavacamten, beyond initial genotyping.
Findings
Variations in the CYP2C19 gene do not explain different clinical outcomes in patients with obstructive hypertrophic cardiomyopathy on mavacamten.
There was a non-significant trend towards faster drug optimization in intermediate and rapid metabolizers compared to normal metabolizers.
Abstract
Mavacamten is the first targeted therapy for obstructive hypertrophic cardiomyopathy (oHCM). It is metabolised via cytochrome p450 enzymes, with variations in the CYP2C19 gene having predominant influence on plasma concentrations of mavacamten. We aimed to outline the effect of CYP2C19 metaboliser status on outcomes in patients taking mavacamten. We retrospectively analysed clinical and echocardiographic data in patients with symptomatic oHCM taking mavacamten. CYP2C19 genotyping was undertaken by loop-mediated isothermal amplification (LAMP) on EDTA whole blood (LaCAR MDx, Liege Belgium) followed by Sanger sequencing of the coding exons of CYP2C19. Logistical regression was used to assess time taken to optimisation. Fifty-five patients (59±13 years; 73% male) were included. Genotyping of CYP2C19*2, CYP2C19*3, and CYP2C19*17 alleles was conducted. Due to low numbers in the ultrarapid…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
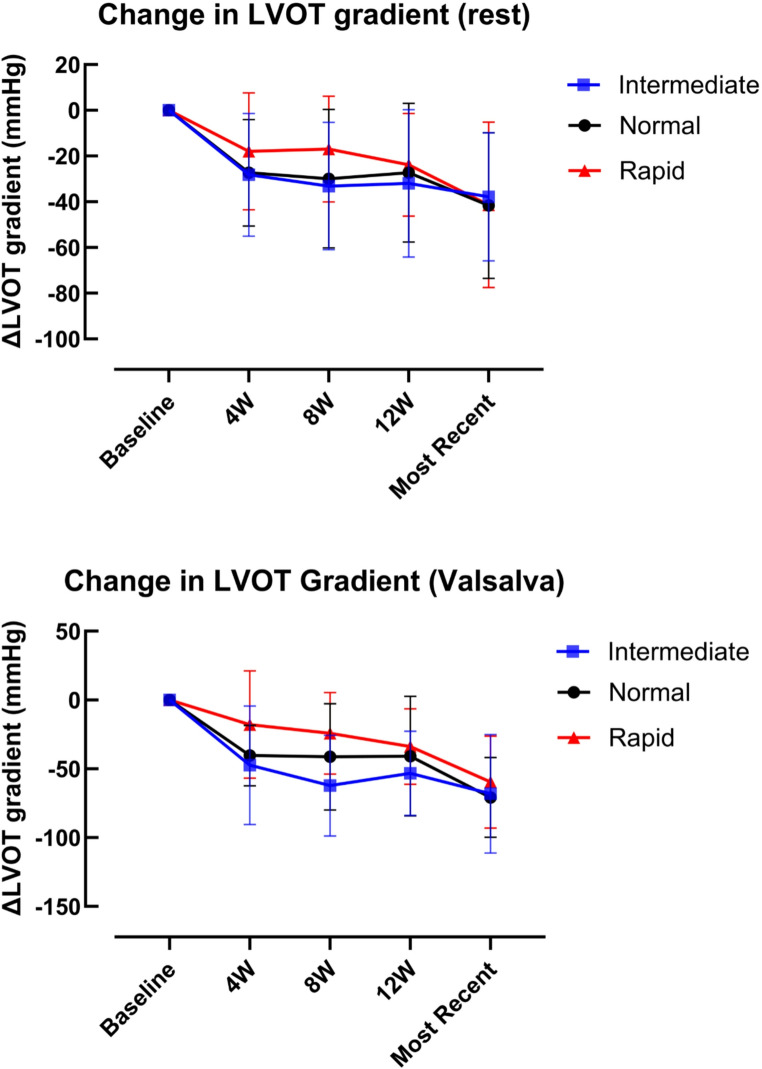 Figure 1
Figure 1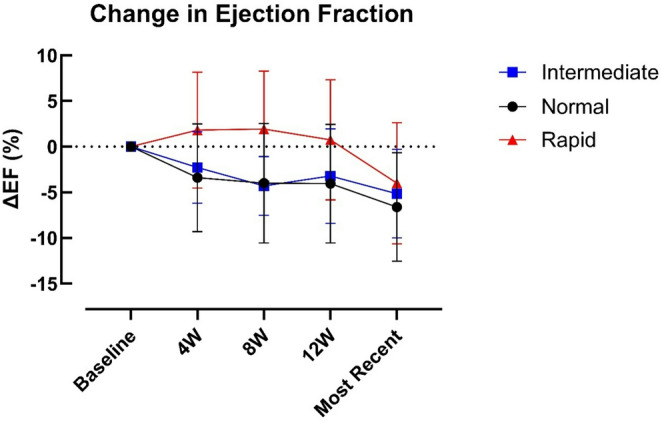 Figure 2
Figure 2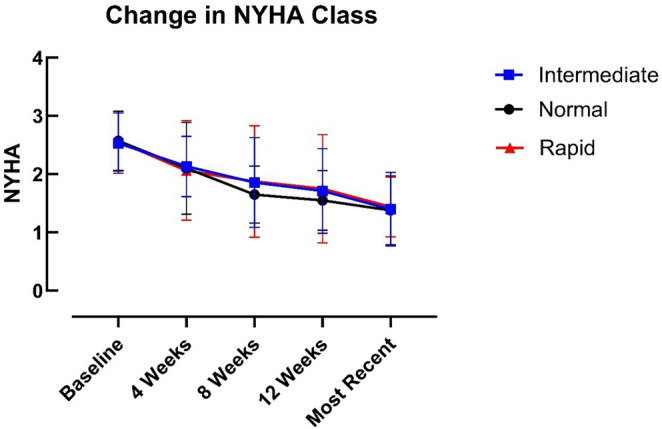 Figure 3
Figure 3Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCardiomyopathy and Myosin Studies · Heart Failure Treatment and Management · Cardiovascular Conditions and Treatments
Introduction
Hypertrophic Cardiomyopathy (HCM) is a condition characterised by abnormal thickening of the left ventricular myocardium. It has a prevalence of at least 1 in 500 individuals and is most commonly caused by mutations in sarcomeric genes [1]. In HCM, there is upregulation of actin-myosin cross bridging in the cardiac myocytes, resulting in a hypercontractile state [2]. Dynamic left ventricular outflow tract (LVOT) obstruction is a core pathophysiological feature of HCM often resulting in symptoms such as chest pain, dyspnoea and exercise limitation [3]. Beta blockers, non-dihydropyridine calcium channel blockers and disopyramide have historically been used to treat LVOT obstruction [4, 5]. However, these non-targeted therapies are often poorly tolerated, with dose titration limited by adverse side effects [6, 7]. Invasive septal reduction therapies such as alcohol septal ablation and surgical myectomy improve long term survival and symptoms in patients with drug-resistant presentations [8]. However, these invasive procedures require centre-specific expertise and may not be appropriate in all patients.
Mavacamten, a first-in class, allosteric inhibitor of cardiac myosin ATPase, targets the underlying hypercontractile physiology of HCM [9]. It is effective in reducing symptoms and improving exercise capacity, as demonstrated in phase III randomised controlled trials [10, 11]. In EXPLORER-HCM, significant reduction in LVOT gradient was seen at 30 weeks, with a 35.6mmHg greater mean reduction in peak post-exercise gradient compared with placebo (95% CI − 43·2 to − 28·1; p < 0·0001). 37% of patients on mavacamten met the primary endpoint, a composite of improved New York Heart Association (NYHA) symptom class and peak oxygen uptake (pVO_2_) (p < 0.0005) [10].
The estimated oral bioavailability for mavacamten is at least 85%, with a rapid median time to maximum concentration (around 1 h) [9]. It is metabolised via the liver, predominantly through cytochrome p450 enzymes CYP2C19, CYP3A4 and CYP2C9. CYP2C19 is responsible for 74% of its metabolism [12, 13]. Individual variants in the CYP2C19 gene lead to variation in mavacamten exposure, with five different metaboliser phenotypes reported: poor, intermediate, normal, rapid and ultrarapid [14]. Elimination half-life varies between these phenotypes: 6 days for ultrarapid metabolisers, 8 days for rapid metabolisers, 9 days for normal metabolisers, 10 days for intermediate metabolisers and 23 days for poor metabolisers [15].
In Europe and the United Kingdom (UK), the summary of product characteristics for mavacamten states that patients should be genotyped for CYP2C19 to determine the appropriate dose. Poor metabolisers or those within unknown metaboliser status start on a lower dose of 2.5 mg rather than 5 mg once daily. The European Medicines Agency outlines a strict dosing regimen, which is separated into a 12 week initiation phase, dose titration and a subsequent maintenance phase. Clinical review with echocardiography occurs at 4 weekly intervals during the first 12 weeks. Dose reduction or temporary cessation of mavacamten occurs if peak LVOT gradient drops to < 20mmHg, or if the left ventricular ejection fraction (LVEF) falls below 50%. Beyond this period, patients are titrated up to a maximal dose of 5 mg (poor metabolisers) or 15 mg in other metaboliser groups, with a target LVOT gradient < 30mmHg and symptom resolution [15]. Currently in the UK, individuals are genotyped for the CYP2C192*, CYP2C193 and CYP2C1917 alleles to determine their metaboliser status. This practice differs from that in North America, where CYP2C19 genotyping is not performed and hence does not inform dosing decisions. Given the time and cost incurred to facilitate genetic testing for these patients, assessment of its utility is an important avenue to explore.
Aims and methods
We sought to determine the effect of CYP2C19 metaboliser status on outcomes in patients with obstructive HCM (oHCM) on mavacamten in two cardiomyopathy centres in the UK.
Study design and setting
Consecutive patients with symptomatic oHCM treated with mavacamten from two UK centres (Guy’s and St Thomas’ Hospitals and Liverpool Heart and Chest Hospital) were included in this retrospective study. Data from baseline, week 4, week 8, week 12 and the most recent visit was collated. This included dose, echocardiographic parameters and New York Heart Association (NYHA) class. Initiation dose was dictated by metaboliser status, with poor metabolisers starting on 2.5 mg and other metaboliser groups starting on 5 mg, as per the European summary of product characteristics.
Patient selection
Patients met established eligibility criteria for mavacamten therapy; including NYHA II-III symptoms, peak LVOT gradient ≥ 50mmHg and left ventricular ejection fraction (LVEF) ≥ 55%. Baseline characteristics are highlighted below in Table 1.
Table 1. Baseline characteristics of patient cohort on MavacamtenN = 55 (SD or %) Age 59 (±13) Sex Male40 (73) Female15 (27) Ethnicity White (British/European)46 (84) Black (African/Caribbean)4 (7) Asian2 (4) Other3 (5) CYP2C19 metaboliser status Ultrarapid1 (2) Rapid16 (29) Normal (extensive)21 (38) Intermediate15 (27) Poor2 (4) HCM Genotype Pathogenic variant in sarcomeric gene10 (18) No pathogenic variant identified33 (60) Variant of unknown significance4 (7) HCM genetic panel not tested/outcome awaited8 (15) Prior HCM treatment Beta blocker46 (84) Non-DHP calcium channel blockers8 (15) Disopyramide24 (44) Previous SRT4 (7) NYHA class at baseline I0 II24 III31 IV0 NYHA at most recent follow up I35 II17 III3 IV0
Genotyping
Patients had CYP2C19 genotyping for the CYP2C192*,* CYP2C193, and CYP2C1917* alleles, undertaken by loop-mediated isothermal amplification (LAMP) on EDTA whole blood (LaCAR MDx, Liege Belgium) as per manufacturer’s instructions at baseline and dosing in the drug initiation phase was determined by this. Poor metabolisers were commenced on 2.5 mg daily, with other phenotypes starting on 5 mg. Sanger sequencing of the nine coding exons and exon/intron boundaries of CYP2C19 (NM_000769.4) was undertaken (primers table S1) on extracted DNA as described previously [16].
Statistical analysis
Patients were considered optimised once treatment entered the maintenance phase and no further dose titration was required, conventionally when the LVOT gradient was < 30mmHg. Descriptive statistics were used to outline baseline characteristics. Logistical regression was performed to assess whether CYP2C19 metaboliser status predicted time to optimisation. Change in ejection fraction (EF) and LVOT gradient was derived using two-way repeated measures ANOVA.
Results
55 patients (59±13 years; 73% male) commenced on mavacamten between December 2023 and September 2024 were included. CYP2C19 metaboliser status was confirmed in the initiation phase (Table 1). Treatment was permanently discontinued in 2 patients due to non-adherence.
Genetics
Genotyping of the CYP2C192*,* CYP2C193, and CYP2C1917* alleles was successfully conducted for all 55 individuals (Table 2). The genotypes are consistent with published allele frequencies [17]. Additional Sanger sequencing of CYP2C19 identified no additional rare or novel variants and the CYP2C19 metaboliser status was not altered for any individual compared to that determined by the genotyping of the three functional alleles.
Table 2. Frequencies of CYP2C19 genotypes and predicted metaboliser statusCYP2C19 GenotypeMetaboliser statusN = 55 (%)CPIC CYP2C19 Approximate genotype frequencies %*1/1Extensive (normal)21 (38)391/2Intermediate11 (20)181/17Rapid16 (29)272/17Intermediate4 (7)62/2Poor2 (4)217/*17Ultrarapid1 (2)4
Time taken to optimisation
44 patients (80%) were optimised by most recent follow up (mean 20.0 ± 12.64 weeks, 95% CI 16.16–23.84). A binary logistic regression model was used to assess whether CYP2C19 metaboliser status predicted delayed optimisation, defined as taking more than 12 weeks to reach target dose or not yet being optimised at last follow-up. Due to low numbers, patients with poor (n = 2) and ultra-rapid (n = 1) phenotypes were excluded. Among the remaining 50 patients, CYP2C19 status (intermediate and rapid vs. normal) was entered as a categorical predictor, using normal metabolisers as the reference.
Intermediate metabolisers had an odds ratio (OR) of 0.63 (95% CI: 0.12–3.19), and rapid metabolisers had an OR of 0.55 (95% CI: 0.11–2.53). The model’s area under the receiver operating characteristic curve (AUC) was 0.57, indicating poor discriminative ability. These findings suggest that CYP2C19 phenotype did not meaningfully predict time to optimisation.
LVOT gradient
Overall reductions in both peak resting (40 ± 34.37 mmHg) and Valsalva (64 ± 35.23 mmHg) gradients were statistically significant (p < 0.0001). Progressive reduction in gradient was observed in rapid, intermediate and normal metabolisers from baseline to the most recent follow-up (Fig. 1). Whilst reduction appeared to be slower in rapid metabolisers, particularly from weeks 4 to 8, this trend was not statistically significant (p = 0.43). There was no interaction between group and time (p = 0.69), indicating that the degree of gradient reduction was similar across all metaboliser types and not influenced by CYP2C19 status.
Fig. 1. Change in LVOT gradient at rest and with Valsalva manoeuvre in normal, intermediate, and rapid CYP metaboliser groups. Data are mean ± SD. Two-way repeated-measures ANOVA with Geisser–Greenhouse correction demonstrated a significant effect of time for both resting and Valsalva gradients (both p < 0.0001), with no effect of CYP metaboliser group (rest p = 0.43; Valsalva p = 0.38) and no time × group interaction (rest p = 0.69; Valsalva p = 0.71)
Rapid reduction in gradients in the initiation phase (< 20mmHg), leading to dose reduction or temporary cessation of mavacamten, was observed in three patients (5%). Two of these patients were normal metabolisers and one was a rapid metaboliser. These patients are now in the maintenance phase of treatment with optimised gradients.
Left ventricular ejection fraction (LVEF)
Post-hoc comparisons demonstrate that in the initiation phase, rapid metabolisers had a marginally smaller reduction in EF at four and eight weeks compared to normal metabolisers (p = 0.04 and 0.03, respectively), and at eight weeks (p = 0.006) compared to intermediate metabolisers (Fig. 2). These differences were transient and resolved by 12 weeks.
Fig. 2. Change in left ventricular ejection fraction (LVEF) during mavacamten therapy across CYP metaboliser groups. Data are shown as mean ± SD. Two-way ANOVA demonstrated significant effects of time (p < 0.0001) and CYP metaboliser group (p = 0.0091), with no time × group interaction (p = 0.21). Tukey post-hoc testing showed higher EF in rapid metabolisers than normal metabolisers at 4 and 8 weeks (p = 0.04 and p = 0.03, respectively) and higher EF in rapid than intermediate metabolisers at 8 weeks (p = 0.006); no other between-group comparisons were significant, and these differences were no longer evident by 12 weeks
One patient had a drop in EF < 50% (intermediate metaboliser) during treatment. LV impairment resolved on mavacamten withdrawal, and the patient has since been established back on mavacamten with optimised gradients and preserved LVEF.
New York heart association class (NYHA)
All patients had NYHA 2–3 symptoms at baseline. Symptoms improved significantly over time (p < 0.0001) in all groups, however there were no significant differences between metaboliser groups at any timepoint (p = 0.63) (Fig. 3). Nearly two-thirds of the group had improved to NYHA 1 at most recent follow up (64%).
Fig. 3. Change in NYHA class from baseline to most recent follow-up in normal, intermediate and rapid metaboliser groups. Data represent median values. Two-way ANOVA showed a significant effect of time (p < 0.0001), with no effect of metaboliser group (p = 0.63) and no time × group interaction (p = 0.99)
Discussion
Our study has shown that CYP2C19 genotyping does not appear to provide additional benefit in predicting response to mavacamten in intermediate, normal and rapid metabolisers.
Establishing CYP2C19 metaboliser status is currently mandated in Europe and the UK. This differs with North America, where genotyping is not part of the dose initiation protocol, and all patients are commenced on 5 mg once daily. Whilst this is the case, there is still an emphasis on close monitoring and consideration of potential drug-drug interactions [18]. Genetic testing incurs additional time and financial resource for genetic and cardiomyopathy services. Clarifying the utility of this is therefore important. Currently, only determination of CYP2C19 poor metaboliser status results in an alteration to mavacamten dose. It is important to establish if different metaboliser status results in altered responses to the drug or identification of other CYP2C19 variants by extended genotyping adds value.
Variants in the CYP2C19 enzyme affect the terminal half-life (t_1/2_) and hence alter drug exposure of mavacamten [9]. There is established data that supports the lower initiation dose in poor metabolisers, with reduced enzyme function leading to higher drug levels and increased risk of adverse events such as LV systolic dysfunction. Whilst the pharmacokinetic profile of mavacamten suggests that identification of poor metabolisers is beneficial, our study was unable to demonstrate this due to low representation from this group. This may be in part related to our demographic of patients (84% White European, see Table 1). Poor metaboliser status varies by ethnicity, with the lowest prevalence seen in the European population (2.1%) and the highest prevalence in Far East Asians (11.9%) [19].
It is worth noting that some of the pharmacokinetic properties of mavacamten may not directly translate to real world practice. Following a single dose of 15 mg mavacamten, AUC increased by 241% and maximum peak concentration increased by 47% in poor metabolisers compared to normal metabolisers [20]. However, as was the case in both EXPLORER and VALOR HCM, the 15 mg dose was proportionally less represented, with only 20% of our cohort established on this in our study [10, 21].
Beyond assessing drug efficacy, predicting which patients may be at increased risk of adverse outcomes is important. In our study, adverse events such as development of LV impairment and rapid reduction in LVOT gradient in the initiation phase were rare, occurred in individuals with different CYP2C19 genotypes, and therefore were not explained by specific variants in the CYP2C19 gene. While the trajectory of LVOT gradient (Fig. 1) and LVEF (Fig. 2) in rapid metabolisers may support a slower effect of the drug in these patients, which is in line with the pharmacokinetic properties of mavacamten, this did not result in different clinical outcomes by most recent follow up (4–60 weeks). Symptomatic improvement was also consistent irrespective of metaboliser status.
Our study has several limitations. Firstly, our small cohort of majority male, White European patients, and the lack of representation from ultrarapid and poor metaboliser groups limits our ability to extrapolate our results to larger, more diverse oHCM populations. Secondly, though we did not identify rare CYP2C19 alleles, our cohort was small and our testing approach would not identify non-coding or structural genetic variants. The low adverse event rate also meant that formal statistical comparisons between CYP2C19 genotypes in this group were not performed. As we could not include the poor metabolisers in our statistical analysis, it is not clear whether CYP2C29 genotyping and subsequent dose amendment in this cohort translates to a better safety profile of the drug. Whilst the elimination half-life of mavacamten (23 days in poor metabolisers compared to 6–10 days for other phenotypes) supports more cautious dosing in this group, presently our data do not support amendments to the UK and European protocol.
Conclusion
Outside CYP2C19 poor metabolisers, variations in the CYP2C19 gene do not appear to effect clinical outcomes in oHCM patients on mavacamten. Whilst a larger and more varied patient cohort is needed to further evaluate this, our study indicates that dose adjustments outside of the CYP2C19 poor metaboliser group are not warranted. Due to small sample size, we were unable to provide definitive evidence as to whether amended dosing in poor metabolisers translates to improved treatment outcomes and a reduction in adverse events. It is therefore unclear whether this practice in the UK and Europe confers any advantage compared to the North American model of care. Nevertheless, the data do not support amendments to the current dosing protocol.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary Material 1
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Medicines and Healthcare Products Regulatory Agency. MHRA products: Camzyos (2024) [Accessed 2025 Jul 14]. Available from: https://mhraproducts 4853.blob.core.windows.net/docs/16c 463c 53f 3ee 48479 bc 36f 92f 021f 3a 813a 1898
- 2European Medicines Agency (EMA). Camzyos (mavacamten): Summary of Product Characteristics (2023) [Accessed 10 Aug 2025]. Available from: https://www.ema.europa.eu/en/documents/product-information/camzyos-epar-product-information_en.pdf
- 3Camzyos (2025) (mavacamten) [prescribing information]. U.S. Food and Drug Administration. Silver Spring (MD): FDA; [Accessed 28 Nov 2025]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2025/214998 s 010lbl.pdf
- 4Full prescribing information [mavacamten] (CAMZYOS™) (2022) [Accessed 2025 Aug 10]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/21499