Chronic Granulomatous Disease: Clinical and Molecular Characterization of Brazilian Patients
Leonardo Martinello da Rosa, Martha Braun da Rosa, Mariana de Sampaio Leite Jobim Wilson, Ida Vanessa Doederlein Schwartz, Fernanda Sperb‐Ludwig

TL;DR
This study characterizes the clinical and genetic features of six Brazilian patients with chronic granulomatous disease, a rare immune disorder.
Contribution
The study identifies novel genetic variants in CYBB and clarifies challenges in variant analysis due to NCF1 pseudogenes.
Findings
Four patients had CYBB variants, including a novel p.Cys257Ser variant affecting the gp91phox protein.
The GT deletion in NCF1 was found in two siblings, highlighting pseudogene-related diagnostic challenges.
Functional studies showed absence of protein expression for the p.Arg157Ter variant.
Abstract
Chronic granulomatous disease (CGD) is a rare inborn error of immunity caused by defects in components of the NADPH oxidase that impair the elimination of infectious microorganisms. Individuals affected by CGD become more susceptible to recurrent and severe infections. Six male patients from Southern Brazil were clinically and genetically analyzed through data collection from medical records and massively parallel sequencing by a panel for the following genes: CYBB, CYBA, NCF1, NCF2, and NCF4 and whole genome sequencing analysis. The gene‐scan technique was used to identify the GT deletion in NCF1. The most common affected organs were the lungs, skin, and lymph nodes; the most common clinical manifestations were recurrent pneumonia, cutaneous involvement, lymph node manifestations, and failure to thrive. Four patients were identified with variants in CYBB: p.Cys257Ser, which is novel;…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
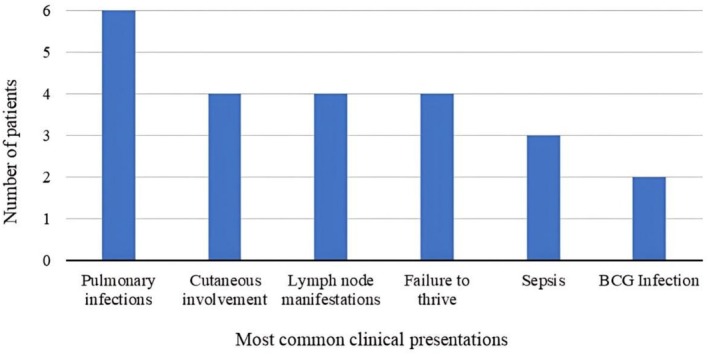 FIGURE 1
FIGURE 1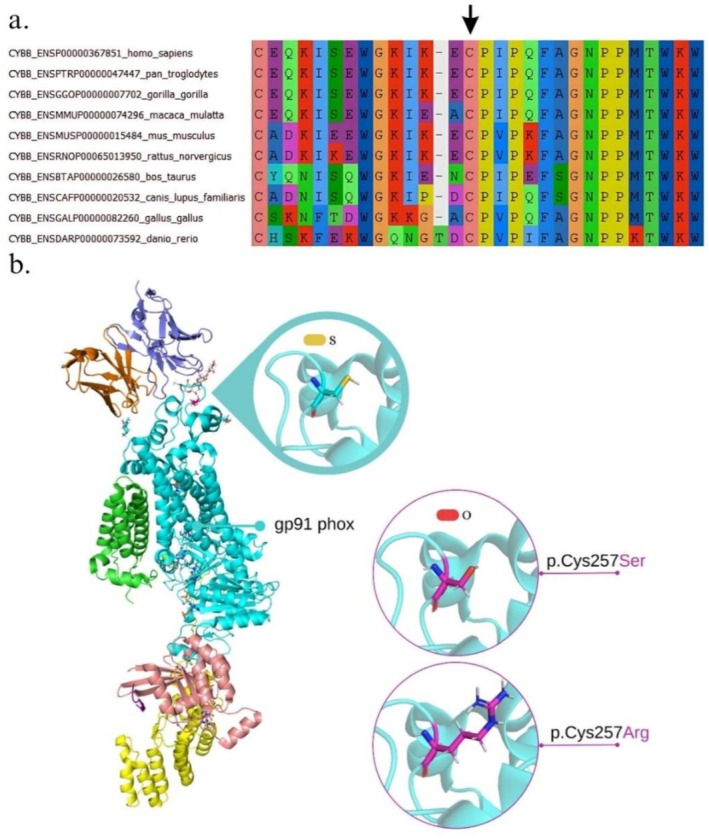 FIGURE 2
FIGURE 2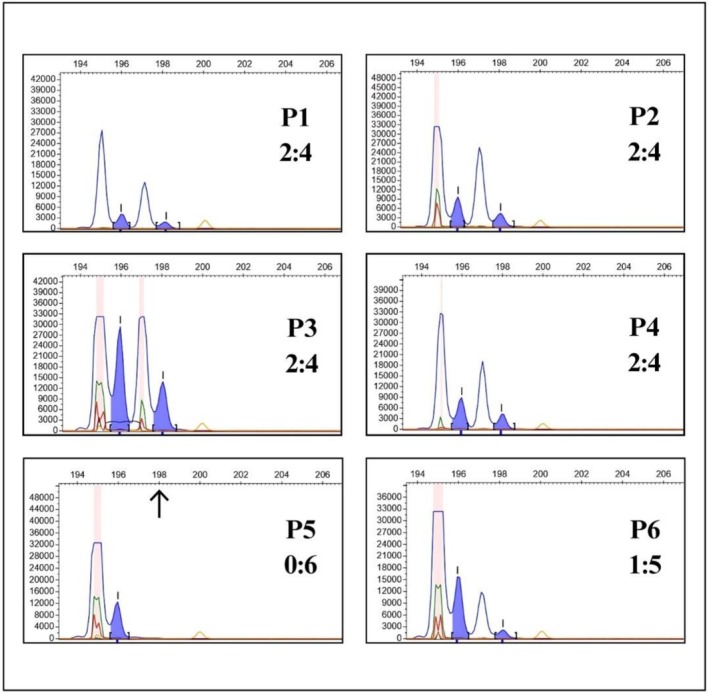 FIGURE 3
FIGURE 3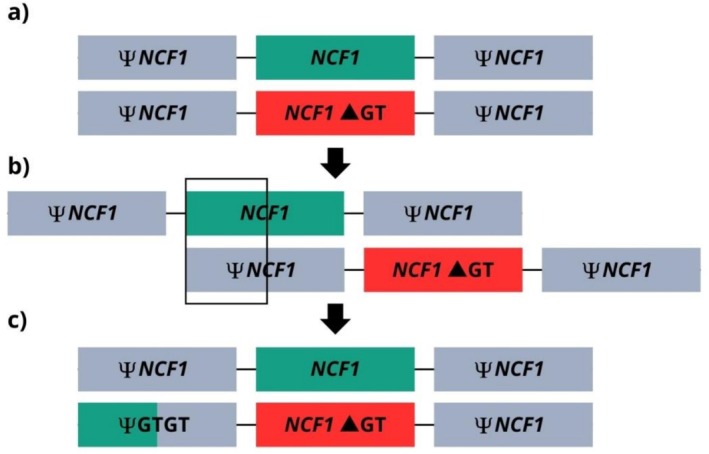 FIGURE 4
FIGURE 4| Cell location | Gene | Protein | Omim | Cytogenetic location | Inheritance | Type of disease |
|---|---|---|---|---|---|---|
| Cell/phagosome membrane |
| gp91phox | 300481 | Xp21.1‐p11.4 | X‐linked | X‐CGD |
|
| p22phox | 608508 | 16q24.2 | Autosomal recessive | CGD4 | |
| Cytoplasm |
| p47phox | 608512 | 7q11.23 | CGD1 | |
|
| p67phox | 608515 | 1q25.3 | CGD2 | ||
|
| p40phox | 601488 | 22q12.3 | CGD3 |
| Patients ( | P1 | P2 | P3 | P4 | P5 | P6 |
|---|---|---|---|---|---|---|
| Sex | Male | Male | Male | Male | Male | Male |
| CGD type | X‐CGD | X‐CGD | X‐CGD | X‐CGD | DCG1 | DGC1 |
| Age of onset | 6y | 4m | 6d | 2m | 8m | 1y 9m |
| Age of clinical diagnosis | 25y | 4y | 6m | 3m | NA | 8y |
| Age of genetic diagnosis | 39y | 11y | 5y | 1y 1m | 6y | 17y |
| Consanguinity | NA | No | No | NA | Yes | Yes |
| Frequent infection | Yes | Yes | Yes | Yes | Yes | Yes |
| First clinical manifestation | Cutaneous lesions | Recurrent pneumonia | Infectious pyoderma | Bronchiolitis | Sepsis | Recurrent pneumonia |
| Granuloma formation | Yes | NA | Yes | Yes | Yes | NA |
| Other clinical manifestations during follow‐up |
BCG infection Bronchopneumonia Chronic lymphadenitis Disseminated fungi infection Lung lesions Lymphadenomegaly Recurrent allergic dermatosis Sepsis |
Auricular abscess Bronchopneumonia Failure to thrive Lymphadenomegaly Pneumocystis pneumonia Sepsis |
Aspergillosis Disseminated BCG infection Bronchiolitis Failure to thrive Hemorrhagic cystitis Lymphadenomegaly Osteomyelitis Recurrent pneumonia Recurrent cutaneous infections and suppurative lesions Sepsis |
Anemia Diarrhea Chronic lymphadenitis Lymphadenomegaly Low Birth Weight Recurrent lymphadenopathy |
Chronic rhinitis Failure to thrive Recurrent cutaneous infections and lesions Respiratory infection Seborrheic dermatitis |
Bronchopneumonia Failure to thrive Recurrent aphthous ulcers Recurrent otitis Allergic dermatosis |
| Detected pathogens |
|
|
| NA | NA |
|
|
IgM (RV) |
118 mg/dL (50–320) |
84 mg/dL (19–146) |
106 mg/dL (3m–1y: 17–150) | NA | NA | NA |
|
IgG (RV) |
2569 mg/d (700–1600) |
741 mg/dL (453–916) |
1178 mg/dL (30d–1y: 203–948) | NA |
1281 mg/dL (2–80y: 540–1822) |
1318 mg/dL (2–80y: 540–1822) |
|
IgA (RV) |
888 mg/dL (100–490) |
166 mg/dL (20–100) |
264 mg/dL (3m–1y: 8–91) | NA |
198 mg/dL (1–11y: 21–291) |
664 mg/dL (34–305) |
|
IgE (RV) |
286 uL/mL (≤ 100) |
25 UI/mL (10–15y: < 200) | NA | NA |
1483 UI/mL (6–9y: < 90) | NA |
|
C‐reactive protein (RV) |
81.60 mg/L (≤ 10.0) |
0.5 mg/dL (< 5.0) |
118.7 mg/dL (< 5.0) | NA |
2.1 mg/dL (< 5.0) |
10.8 mg/dL (< 5.0) |
|
CD3+ (RV) |
920.4 uL (19–44y: 844–1943) |
3615.6 uL (2–6y: 1515–3701) |
5071 uL (6–12m: 2153–5004) | NA | NA |
1333 uL (1045–2760) |
|
CD3+/CD4+ (RV) |
542.8 uL (19–44y: 476–1136) |
2449.7 uL (2–6y: 618–1348) |
3538 uL (6–12m: 1360–3066) | NA | NA |
61 uL (550–1680) |
|
CD3+/CD8+ (RV) |
220.7 uL (19–44y: 248–724) |
828.7 uL (2–6y: 453–1700) |
1250 uL (6–12m: 560–1803) | NA | NA |
599 uL (285–1175) |
|
CD19+ (RV) |
88.5 uL (19–44y: 138–544) |
926.1 uL (2–6y: 931–1283) |
1453 uL (6–12m: 811–1792) | NA | NA |
427 uL (160–600) |
|
CD3‐/CD16+56+ (RV) |
171.1 uL (19–44y: 134–545) |
754.4 uL (2–6y: 135–601) |
726 uL (6–12m: 164–801) | NA | NA |
122 uL (110–910) |
|
DHR‐Assay Ref. 1 |
NS: 1.80 (C:0.70) WS: 38.50 (C:95.30) |
NS: 0.20 (C:0.30) WS: 49.30 (C:84.30) |
NS: 0.10 (C:0.40) WS: 0.10 (C:99.40) |
NS: 0.10 (C:0.80) WS: 0.30 (C:99.30) | NA |
NS: 0.10 (C:0.20) WS: 50.70 (C:89.70) |
|
NBT‐Assay Ref. 2 |
NS: 1% WS: 5% |
NS: 1% WS: 2% | NA | NA | NA |
NS: 2% WS: 3% |
| HSCT | NA | NA | BMT | NA | NA | BMT |
| Current age | 39y | 11y | 5y | 1y10m | 6y | 17y |
| Patients ( | P1 | P2 | P3 | P4 | P5 | P6 |
|---|---|---|---|---|---|---|
| Affected gene |
|
|
|
|
|
|
| Protein | gp91phox | gp91phox | gp91phox | gp91phox | p47phox | p47phox |
| Variant type | Missense | Missense | Nonsense | Nonsense | Frameshift | Frameshift |
| Nucleotide (c.) | c.769T>A | c.769T>C | c.469C>T | c.1449G>A | c.75_76delGT | c.75_76delGT |
| Amino acid (p.) | p.Cys257Ser | p.Cys257Arg | p.Arg157Ter | p.Trp483Ter | p.Tyr26HisfsTer | p.Tyr26HisfsTer |
| Location | Exon 7 | Exon 7 | Exon 5 | Exon 11 | Exon 2 | Exon 2 |
| Zygosis | Hemizygous | Hemizygous | Hemizygous | Hemizygous | Heterozygous | Homozygous |
| ACMG criteria | PM1 PM2 PM3 PP3 | PM1 PM2 PM3 PP3 | PVS1 PM3 PP3 | PVS1 PM3 PP3 | PVS1 PM1 PM2 PM4 PP3 | PVS1 PM1 PM2 PM4 PP3 |
| Classification | Likely pathogenic | Likely pathogenic | Pathogenic | Pathogenic | Pathogenic | Pathogenic |
|
| 1787/4070 | 4445/9735 | 13,947/29,494 | 4252/8980 | Abs./12,569 | 2222/15,754 |
|
| 2:4 (0.44) | 2:4 (0.46) | 2:4 (0.47) | 2:4 (0.47) | 0:6 (0) | 1:5 (0.14) |
| Interaction energy | (ΔΔG = ΔGmut − ΔGwt) | ||
|---|---|---|---|
| Chains | Wild type | p.Cys257Ser | p.Cys257Arg |
| AB | −35.38 | 0 | 0 |
| BC | 0 | 0 | 0 |
| BD | −30.82 | 0.55 | 0.64 |
| BE | −16.58 | 2.43 | 0.15 |
| BH | −2.47 | 1.23 | 0.03 |
| BL | −0.49 | −0.75 | −0.79 |
- —Fundação de Amparo à Pesquisa do Estado do Rio Grande do Sul10.13039/501100004263
- —Fundo de Incentivo à Pesquisa e Eventos do Hospital de Clínicas de Porto Alegre
- —Conselho Nacional de Desenvolvimento Científico e Tecnológico10.13039/501100003593
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsNeutrophil, Myeloperoxidase and Oxidative Mechanisms · Immunodeficiency and Autoimmune Disorders · Asthma and respiratory diseases
Introduction
1
Chronic granulomatous disease (CGD) is a rare inborn error of immunity (IEI), with an overall estimated incidence of 1 in every 250,000 live births, caused by defects in the components of nicotinamide adenine dinucleotide phosphate oxidase complex (NADPH oxidase). The complex is present in phagocytic cells and is responsible for producing reactive oxygen species (ROS), which are essential for the elimination of pathogens. The loss or decrease in ROS production due to defects in the NADPH oxidase compromises its microbicidal function [1, 2]. Affected individuals become more susceptible to bacterial and fungal infections, inflammatory complications, autoimmunity, and malignancies and are commonly infected by a narrow spectrum of pathogens, typically catalase producers. The most common pathogens reported in North America and Europe infecting patients with CGD include Staphylococcus aureus and species of the genera Serratia, Nocardia, Burkholderia, Salmonella, Candida, and Aspergillus, Bacillus Calmette‐Guérin (BCG), and Mycobacterium tuberculosis are also relevant pathogens in developing countries, where tuberculosis is endemic and/or BCG vaccine is routinely given, as in Brazil [3, 4, 5, 6].
The five protein components of the NADPH oxidase are encoded by five genes. Thus, a genetic defect in any of the genes disrupts the corresponding protein and impairs the overall function of the complex, leading to the main forms of CGD: the X‐linked recessive form (X‐CGD), caused by defects in CYBB gene (gp91^phox^ protein), and the autosomal recessive forms (AR‐CGD), caused by defects in the following genes: NCF1 (p47^phox^), NCF2 (p67^phox^), and NCF4 (p40^phox^), and CYBA (p22^phox^) [7, 8].
Diagnosis of CGD is based on the identification of clinical presentations along with assessment of NADPH oxidase activity through measurement of ROS production, followed by genetic testing, which is used to confirm the exact affected gene. The nitroblue tetrazolium (NBT) and dihydrorhodamine 123 (DHR) tests are widely used to diagnose the disease. However, these tests do not determine the specific genetic form of CGD and may yield false‐positive results related to other conditions where NADPH oxidase activity is also impaired. In this context, genetic testing becomes essential not only for genetic counseling and confirmation of the specific type of CGD but also for the identification and referral of individuals for hematopoietic stem cell transplantation (HSCT), the most effective curative therapy for patients with CGD [1, 5, 9, 10]. In particular, diagnosing CGD1 (involving the NCF1 gene) can be challenging when only based on traditional sequencing methods because NCF1 has two nonfunctional pseudogenes (ΨNCF1), which share 99% sequence homology with the functional gene. One of the few distinguishing features among them is a GT deletion (ΔGT) at the beginning of exon 2, naturally present in ΨNCF1, which can also arise in functional NCF1 alleles, resulting in the frameshift variant p.Tyr26HisfsTer [11].
Despite numerous advances in recent decades, the diagnostic process for IEIs, including CGD, remains challenging. Many patients still undergo a diagnostic odyssey without reaching a definitive conclusion. This has a significant impact on their quality of life, delaying both preventive care and potential treatments [12, 13].
Given the clinical relevance of CGD, the scarcity of studies correlating clinical and genetic aspects of the disease, and the need to establish efficient genetic and molecular diagnostic protocols in a genetically diverse population, we characterized a cohort of Brazilian patients through clinical and genomic analyses. We identified the causative genes and variants, performed structural molecular analyses, and described for the first time the p.Cys257Ser variant in the CYBB gene and proposed the hypothesis of genotype resulting from recombination events involving NCF1 and its pseudogenes.
Materials and Methods
2
This study was approved by the Research Ethics Committee of the Hospital de Clínicas de Porto Alegre (CEP‐HCPA FIPE 2022‐0206). Written informed consent was obtained from all participants or their legal guardians. Six male patients from Southern Brazil with clinical suspicion of CGD had revised their clinical information from medical records and blood samples collected in EDTA vacuum tubes; genomic DNA was extracted using the Easy‐DNA Purification Kit (Thermo Fisher) and quantified using the NanoDrop 1000 spectrophotometer (Thermo Fisher). A custom gene panel was designed using the Ion AmpliSeq Designer software (Thermo Fisher) and included the following genes: CYBB, CYBA, NCF1, NCF2, and NCF4 (Table 1). Sequencing was performed on the Ion Torrent PGM platform (Thermo Fisher) with a minimum coverage of 150×. Data processing was conducted using Torrent Suite v5.0.5, and the GRCh37.p13 assembly was used as the reference genome. Whole genome sequencing was performed on the MGI PCR‐free platform and processed with the Sentieon Germline Pipeline v1.0, using the GRCh38 assembly as the reference genome, with a mean coverage of 30×. The gene‐scan method was employed according to Dekker et al. [11] in order to identify the ΔGT and distinguish the NCF1 gene from its pseudogenes (ΨNCF1). The fragments were separated by capillary electrophoresis using the ABI 3500 Genetic Analyzer (Applied Biosystems), with the GS500(‐250)LIZ size standard. Electropherogram analysis and peak height identification for NCF1 and ΨNCF1 were performed using Microsatellite Analysis software on the Thermo Fisher Cloud platform. Variant analysis was performed using the following tools: Variant Effect Predictor [14], Ion Reporter (Thermo Fisher), and Integrative Genomics Viewer [15]. Variant classification followed the criteria of the American College of Medical Genetics and Genomics (ACMG) [16] and the Clinical Genome Resource [17].
The amino acid sequences of gp91^phox^ from 10 different species were retrieved from the Ensembl database [18] to perform a multiple sequence alignment, which was carried out using the MUSCLE software [19]. The three‐dimensional structure of the NADPH oxidase was obtained from the Research Collaboratory for Structural Bioinformatics Protein Data Bank (RCSB PDB) under the identifier 8WEJ [20]. Structural modeling and image generation of the p.Cys257Arg and p.Cys257Ser variants were conducted using PyMOL software [21]. Both wild‐type and mutant NADPH oxidase structures were submitted to structural repair using FoldX v5.0 [22]. Gibbs free energy (ΔG) values were calculated for the overall structure and for interchain interactions in the wild‐type (ΔGwt) and mutant (ΔGmut) NADPH oxidase complexes; differences in ΔΔG (ΔGmut − ΔGwt) greater than +1.6 kcal/mol were considered indicative of significant alterations to the complex [23, 24].
Results
3
Six male patients were analyzed. Clinical and biochemical information are shown in Table 2, and the genetic information is shown in Table 3. The mean age at symptom onset was approximately 18 months (6 days—6 years). The mean age at the clinical diagnosis was 7.5 years (n = 5; 3 months—25 years). The mean age at the genetic diagnosis was 13.1 years (n = 6; 1–39 years). The most common manifestations involved recurrent infections mainly on the lungs, observed in all patients, including bronchopneumonia, bronchiolitis, pneumocystis pneumonia, and recurrent pneumonia, and on the skin, observed in 85% of the patients (n = 4), including allergic dermatosis, seborrheic dermatitis, infectious pyoderma, and other cutaneous infections (Figure 1). The lymph nodes were also commonly affected, with manifestations occurring in 85% of the patients (n = 4), including lymphadenitis, lymphadenomegaly, and recurrent lymphadenopathy.
Most common clinical presentations observed in patients P1–P6.
Four patients were identified with alterations in the CYBB gene with four different variants, one of which (p.Cys257Ser) is novel in the literature (P1; Table 3). Multiple sequence alignment across 10 different species demonstrated that the cysteine residue at position 257, where both missense variants in gp91^phox^ occur (p.Cys257Ser and p.Cys257Arg), is highly conserved (Figure 2a). The amino acid substitutions caused by these variants in gp91^phox^ are illustrated in Figure 2b; structural analyses of the NADPH complex for the missense variants revealed significant changes in binding affinity energy (ΔΔG) between chains B (gp91^phox^) and E (a small GTPase) in the p.Cys257Ser variant, as well as in the overall structural stability (ΔΔG_total_) of both variants (Table 4).
Three‐dimensional modeling of NADPH oxidase and multiple sequence alignment for a cysteine residue in gp91phox across 10 different species. (a) Multiple sequence alignment of the gp91phox amino acid sequence across 10 different species using MUSCLE v3.8, revealing the high conservation of the Cys257 residue (indicated by the arrow). (b) Structural representation of NADPH oxidase, showing the wild‐type cysteine residue in the position 257 in gp91phox subunit and the two missense variants identified in this study (highlighted in magenta).
The gene‐scan results are shown in Figure 3. The analysis revealed the p.Tyr26HisfsTer (ΔGT) variant in NCF1 in homozygous form in patient P5 and in heterozygous form in patient P6, possibly due to a ΨNCF1 lacking the ΔGT, which may have arisen from unequal recombination event (Figure 4). After whole genome sequencing analysis performed in P6, no additional variants in CGD‐associated genes were identified.
Fragment analysis by the gene‐scan technique of patients P1–P6. The blue peaks on the left correspond to ΨNCF1 (196 bp), and the blue peaks on the right correspond to NCF1 (198 bp). P1–P4 had ratios close to 0.50, reflecting a 2:4 proportion of NCF1/ΨNCF1, considered normal values in individuals without ΔGT in the functional NCF1 gene; P5 had a ratio of zero, indicating homozygosity for the ΔGT in functional NCF1 gene (arrow indicates absence of functional NCF1 alleles). P6 had a ratio of 0.14 and a 1:5 proportion, consistent with a heterozygous ΔGT in one functional NCF1 allele.
A possible recombination event between NCF1 and ΨNCF1 as a hypothesis for the observed heterozygous ΔGT profile in P6. (a) Both parents of P5 and P6 are likely heterozygous for the ΔGT, as supported by the homozygous genotype observed in P5. (b) An unequal recombination event, previously described by Hayrapetyan et al. [25], may have occurred in one parent, in which NCF1 and ΨNCF1 exchanged their first segment, leading to the insertion of the ΔGT into functional NCF1 alleles and, simultaneously, the insertion of the normal GTGT sequence into ΨNCF1. (c) A possible explanation for the genotype of P6 is the inheritance of a ΨNCF1 carrying the GTGT sequence from one parent, which could confound the analyses by being interpreted as a functional NCF1 sequence. Adapted from [25].
Discussion
4
This is the first genetic study conducted in a cohort with CGD patients from Southern Brazil. Previous studies in Brazil have been mostly concentrated in the Southeast region, which harbors one of the most admixed populations worldwide. In contrast, the population of Southern Brazil is characterized by a predominantly European ancestry, accounting for approximately 80% of its genetic background [26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36].
In general, lymphadenitis is the first clinical manifestation observed in patients with CGD. However, in the present study, the first clinical manifestations involved pulmonary infections in 50% of the patients (n = 3), cutaneous infections in 33% (n = 2), and sepsis in 16% (n = 1). Pneumonia is the most common clinical manifestation observed in CGD, affecting 70%–80% of affected patients, and all the patients analyzed were affected by recurrent pneumonia. In this cohort, the organs most frequently affected by infections were the lungs, skin, and lymph nodes, consistent with the literature [8, 10, 37, 38, 39, 40, 41, 42]. Granuloma formation is a typical feature of CGD, which occurs due to excessive inflammatory response, and it was registered in 66.67% (n = 4) of the analyzed patients. Genetic variation among innate immunity molecules may contribute to differences in the individual inflammatory response and their severity, modifying the risk of developing inflammatory complications in patients with CGD [6].
Variants in CYBB account for more than 60% of all reported CGD cases, representing the leading cause of the disease and predominantly affecting males. Variants in NCF1 contribute to approximately 20% of cases and represent the most frequent cause of AR‐CGD; variants in CYBA and NCF2 are responsible for about 14% of cases [2]. Among the patients analyzed, 66.67% (n = 4) were diagnosed with X‐CGD caused by CYBB variants, and 22.23% (n = 2) with AR‐CGD due to the ΔGT in NCF1. Only 50% of these patients (n = 3) had previously received a clinical diagnosis based on biochemical testing, but without a definitive classification of the CGD subtype.
The most severe cases of CGD are associated with extremely low or absent ROS levels, usually resulting from nonsense CYBB variants that cause complete loss of gp91^phox^ function and tend to present earlier in life. Such was the case of patient P3, who developed symptoms at 6 days of life, representing the most severe phenotype among those analyzed. In contrast, AR‐CGD cases in general, as well as some X‐linked cases caused by missense variants affecting amino acids 1–309 (except residue 222) of gp91^phox^, are associated with better survival. These patients usually present with milder to moderate phenotypes due to residual ROS production, which can cause a delayed onset of symptoms as well as a delay in definitive diagnosis, contributing to the overall low diagnosis rate [1, 7, 8, 12, 43].
Most patients with CGD are diagnosed before the age of five. However, in the present cohort, only one patient (P4) received both clinical and genetic diagnoses within this age range. Late diagnosis is common among Brazilian patients, with reported ages at diagnosis ranging from 8 to 19 years [28, 32, 35, 36, 38, 40, 44, 45].
Two patients (P1 and P3) had infections resulting from BCG vaccination, P3 with the disseminated form. Complications related to BCG vaccination are commonly observed in patients with CGD, due to their inability to eliminate the attenuated Bacillus. Data from Latin America and Asia show that such complications occur in 11%–58% of patients with CGD and may represent the first clinical manifestation of the disease. Although rare, disseminated BCG infection is associated with a mortality rate of 60%–80%. In Brazil, the BCG vaccine is routinely administered to newborns and children before 5 years of age because of tuberculosis endemism in the country. Therefore, it is crucial to contraindicate the BCG vaccination for patients with CGD [3, 30, 40, 41, 44].
Genetic testing plays a key role in the efficient and early determination of the specific type of CGD, contributing to appropriate disease management and genetic counseling, as well as supporting the selection and referral of patients for HSCT [5, 7, 10, 39, 46].
Structural alterations in gp91^phox^ can affect the electron transfer site from NADPH to O_2_ in extra‐ or intracellular compartments, thereby determining ROS production [47]. Two patients diagnosed with X‐CGD had missense variants in the CYBB gene, in which the p.Cys257Ser variant identified in patient P1 is a novel finding in the literature and was classified as likely pathogenic. This patient was clinically and genetically diagnosed at the ages of 25 and 39, respectively. Barkai et al. [48] also reported CGD patients with CYBB missense variants in Israel with delayed diagnosis, with a mean diagnostic age of 57.3 years. In patient P2, the likely pathogenic variant p.Cys257Arg was identified, which was previously described in Japanese and Chinese patients. Although BCG infections were identified in the Japanese cohort, no genotype correlation was observed, and such infections were not reported in P2 [49, 50, 51].
The Cys257 residue in gp91^phox^ affected by CYBB missense variants is located in loop E, the largest among the loops of gp91^phox^ (loops A and C), a region of structural and functional relevance. Loop E also contains an N‐linked glycosylation site essential for protein folding, stability, and function. In addition, this loop stabilizes and correctly positions an external heme group of the gp91^phox^, which is crucial for electron transfer and ROS generation. Furthermore, residues Cys257 and Cys244 form a disulfide bond within gp91^phox^, which plays a fundamental role in maintaining tertiary and quaternary conformation of proteins. Both variants involving the Cys257 residue were previously predicted by Noreng et al. [52] to disrupt the structure of loop E. In addition, this loop is highly conserved among mammals as well as this cysteine residue, which is conserved across 10 species, emphasizing its biological and evolutionary significance [20, 46, 53, 54, 55, 56].
The ΔΔG_total_ values for both variants observed in patients P1 and P2 revealed a stability loss in the overall structure of the NADPH oxidase, suggesting a significant functional impact; ΔΔG values greater than 3 kcal/mol indicate a high destabilization in the protein structure. In addition, ΔΔG values of interchain affinity energies in B and E chains of p.Cys257Ser were found to be significant, suggesting an impact on the interaction between gp91^phox^ and a small GTPase (RAC1/2), which may affect the activation of the NADPH oxidase [20, 23, 47, 57, 58].
The nonsense variant p.Arg157Ter in CYBB, identified in patient P3, was classified as likely pathogenic and is reported in more than 60 patients across Latin America, North America, Asia, and Europe [41, 59, 60, 61, 62, 63, 64, 65, 66]. This variant occurs within a CpG island involved in transcriptional regulation through cytosine demethylation, which is considered a mutational hotspot in CYBB. Moreover, it was previously reported in Brazil, where the absence of gp91^phox^ expression was demonstrated in a patient [26, 31].
In patient P4, the pathogenic variant p.Trp483Ter in CYBB was identified. This variant has been reported in studies from India and Denmark, with the respective patients receiving a diagnosis before the age of 5, as was the case with P4 [64, 67]. The Indian patient reported by Rawat et al. [64] died within the first year of life, whereas the Danish patient presented with chronic urticaria and cerebral abscesses. This variant is predicted to undergo nonsense‐mediated mRNA decay (NMD), a mechanism that targets transcripts harboring a premature termination codon located more than 50 nucleotides upstream of the last exon–exon junction, thereby preventing accumulation of nonfunctional transcripts and aberrant proteins, leading to the absence of gene products [68]. In addition, alterations occurring beyond residue 310 of gp91^phox^ impair the binding domains for FAD and NADPH, which are crucial for ROS production [7, 46].
The p.Tyr26HisfsTer (ΔGT) variant in NCF1, identified in patients P5 and P6, accounts for more than 90% of all AR‐CGD cases and leads to loss of function of p47^phox^. Due to the high homology between NCF1 and its pseudogenes, several hotspots for recombination events exist, which can occur unequally and insert the ΔGT in NCF1, which is estimated to occur in 1 out of every 250 individuals. Patients P5 and P6 are siblings from consanguineous parents. P5 is homozygous for ΔGT, whereas P6 was identified as heterozygous by gene‐scan, gene panel, and whole genome analysis. Despite presenting a clinical phenotype of CGD, no additional variants were detected in NCF1 or in the other CGD‐associated genes in P6. A possible explanation for this heterozygous profile is the presence of a ΨNCF1 carrying the GTGT sequence instead of the ΔGT, as previously described by Heyworth et al. [69] and Hayrapetyan et al. [25], which may have arisen through unequal recombination with NCF1. Considering that the parents are consanguineous and likely heterozygous for ΔGT, as indicated by the homozygosity observed in P5, it is plausible that a recombination event occurred during gametogenesis, leading to the observed genotype discrepancy between the siblings (Figure 4). Moreover, the high degree of homology between NCF1 and its pseudogenes complicates sequencing analysis, frequently resulting in alignment errors, misreads, and incorrect variant calls. Furthermore, unequal recombination events can cause deletion of entire gene segments, which are also difficult to detect using conventional sequencing methods or even the gene scan [2, 11, 70, 71, 72].
Conclusions
5
CGD belongs to a group of rare and underdiagnosed diseases, highlighting the importance of characterizing clinical presentations and establishing a genetic diagnosis through genotype–phenotype correlations. Beyond guiding appropriate patient management and genetic counseling, genetic diagnosis contributes to the referral of patients for HSCT and enables the contraindication of BCG vaccination, thereby preventing adverse and potentially life‐threatening reactions. In the present study, it was possible to document both the clinical and genetic heterogeneity of Brazilian patients with CGD, as well as to identify and characterize a novel variant causing the disease.
Author Contributions
Leonardo Martinello da Rosa: data curation, formal analysis, investigation, methodology, project administration, software, validation, visualization, writing – original draft, writing – review and editing. Martha Braun da Rosa: formal analysis, investigation, methodology, software, visualization, writing – review and editing. Mariana de Sampaio Leite Jobim Wilson: conceptualization, data curation, investigation, methodology, resources, validation, writing – review and editing. Ida Vanessa Doederlein Schwartz: conceptualization, data curation, funding acquisition, methodology, project administration, resources, supervision, writing – review and editing. Fernanda Sperb‐Ludwig: conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, project administration, resources, software, supervision, validation, visualization, writing – original draft, writing – review and editing.
Funding
This work was supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico; Fundação de Amparo à Pesquisa do Estado do Rio Grande do Sul (FAPERGS 24/2551‐0000591‐9), and Fundo de Incentivo à Pesquisa e Eventos do Hospital de Clínicas de Porto Alegre (2022‐0206).
Ethics Statement
This study was approved by the Research Ethics Committee of Hospital de Clínicas de Porto Alegre under protocol number 2022‐0206, approved on December 28, 2022, in conformity with the principles of the Declaration of Helsinki.
Consent
Written informed consent was obtained from all participants or their legal guardians prior to inclusion in the present study.
Conflicts of Interest
The authors declare no conflicts of interest.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1C. J. O'donovan , L. T. Tan , M. A. Z. Abidin , M. R. Roderick , A. Grammatikos , and J. Bernatoniene , “Diagnosis of Chronic Granulomatous Disease: Strengths and Challenges in the Genomic Era,” Journal of Clinical Medicine 13, no. 15 (2024): 4435, 10.3390/jcm 13154435.39124702 PMC 11313294 · doi ↗ · pubmed ↗
- 2D. Roos , K. Van Leeuwen , A. P. Hsu , et al., “Hematologically Important Mutations: The Autosomal Forms of Chronic Granulomatous Disease (Third Update),” Blood Cells, Molecules, and Diseases 2 (2021 a): 102596, 10.1016/j.bcmd.2021.102596.34547651 · doi ↗ · pubmed ↗
- 3Brasil Ministério da Saúde , “Boletim Epidemiológico – Tuberculose 2025: Número Especial, Março de 2025. Brasília: Ministério da Saúde, Secretaria de Vigilância em Saúde e Ambiente, Departamento de HIV, Aids, Tuberculose, Hepatites Virais e IST,” (2025), accessed May 28, 2025, https://www.gov.br/saude.
- 4F. Conti , S. O. Lugo‐Reyes , L. B. Galicia , et al., “Mycobacterial Disease in Patients With Chronic Granulomatous Disease: A Retrospective Analysis of 71 Cases,” Journal of Allergy and Clinical Immunology 138, no. 1 (2016): 241–248, 10.1016/j.jaci.2015.11.041.26936803 · doi ↗ · pubmed ↗
- 5A. A. Justiz‐Vaillant , A. F.‐A. Williams‐Persad , R. Arozarena‐Fundora , et al., “Chronic Granulomatous Disease (CGD): Commonly Associated Pathogens, Diagnosis and Treatment,” Microorganisms 11, no. 9 (2023): 2233, 10.3390/microorganisms 11092233.37764077 PMC 10534792 · doi ↗ · pubmed ↗
- 6M. G. Schäppi , V. Jaquet , D. C. Belli , and K.‐H. Krause , “Hyperinflammation in Chronic Granulomatous Disease and Anti‐Inflammatory Role of the Phagocyte NADPH Oxidase,” Seminars in Immunopathology 30, no. 3 (2008): 255–271, 10.1007/s 00281-008-0119-2.18509648 · doi ↗ · pubmed ↗
- 7J. W. Leiding and S. M. Holland , “Chronic Granulomatous Disease,” in Gene Reviews, eds. M. P. Adam , J. Feldman , G. M. Mirzaa , et al. (University of Washington, 2022), 1–352.22876374 · pubmed ↗
- 8H.‐H. Yu , Y.‐H. Yang , and B.‐L. Chiang , “Chronic Granulomatous Disease: A Comprehensive Review,” Clinical Reviews in Allergy & Immunology 61, no. 2 (2020): 101–113, 10.1007/s 12016-020-08800-x.32524254 · doi ↗ · pubmed ↗