Striking a balance: how the gut microbiome shapes the fate of intestinal CD4+ T cells
Jessica M Till, Orion D Brock, Philip P Ahern

TL;DR
The gut microbiome influences immune tolerance by shaping intestinal CD4+ T cells, which is crucial for maintaining a balanced immune response.
Contribution
This paper reviews how the gut microbiome shapes intestinal CD4+ T cells and highlights gaps in leveraging these interactions for clinical applications.
Findings
The gut microbiome and immune system co-evolved to promote immunological tolerance.
Specific microbiome members influence immune function, but the mechanisms remain poorly understood.
CD4+ T cells are essential for maintaining tolerance, yet clinical translation remains challenging.
Abstract
The induction of immune tolerance, a state of immunologic hyporesponsiveness to an antigen, is essential to prevent the destructive potential of the immune system in response to harmless or beneficial agents. Early efforts to understand tolerance focused on model stimuli, self-antigens, transplanted organs, and the growing fetus. Through co-evolution, the microbiome and the host immune system have developed strategies that promote immunological tolerance to the microbiome. This dialogue ensures the maintenance of mutualistic interactions that provide a stable habitat for the microbiome which in turn confers numerous physiological benefits to the host. Despite the gut microbiome being a potent inducer of immune tolerance, the mechanisms through which specific members shaped immune function remained largely ignored for decades. The growing appreciation for the immunomodulatory capacity of…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
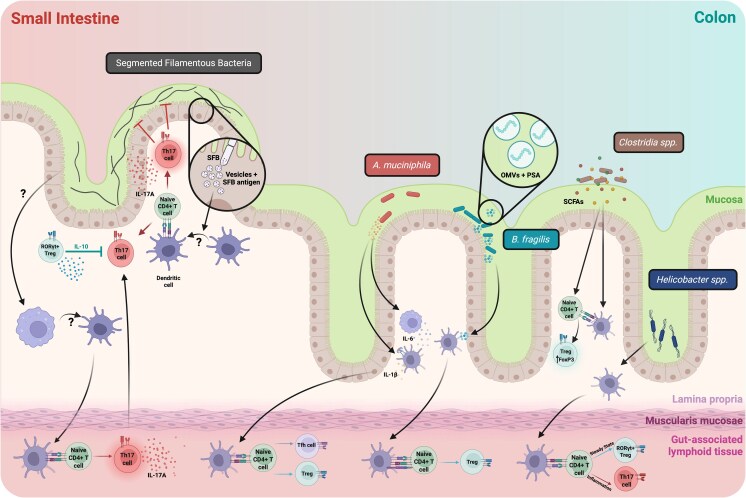 Figure 1
Figure 1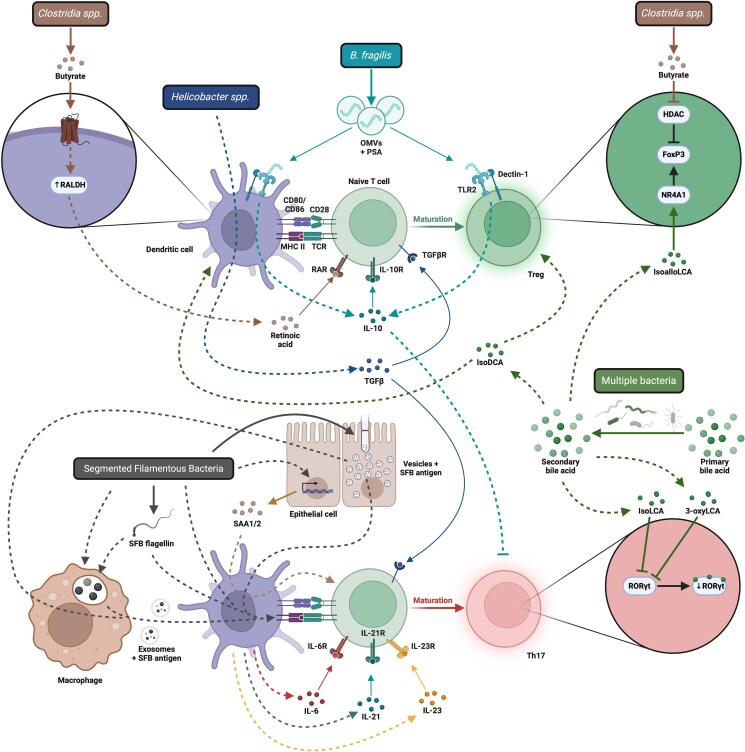 Figure 2
Figure 2- —National Institute of Diabetes and Digestive and Kidney Diseases10.13039/100000062
- —National Institute of Allergy and Infectious Diseases10.13039/100000060
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGut microbiota and health · T-cell and B-cell Immunology · Immune responses and vaccinations
Introduction
Foundational studies describing the generation of immunological tolerance by Owen, Burnet, Medawar and others inspired immunologists to understand the mechanistic underpinnings of this phenomenon, in hopes of gaining insights into the etiology of autoimmune disease and providing novel avenues for the treatment of immune-driven disorders [1–5]. Nowhere are the challenges associated with the induction of tolerance more evident than the intestine, a unique immunologic site home to a vast collection of bacterial, fungal, viral, and archaeal agents, collectively referred to as the microbiome, as well as a dense network of immune cells. Given the potential for these immune cells to generate tissue-destructive responses in response to stimulation with microbial products, it is essential that the microbes (used here to include viruses) that make up the microbiome and the intestinal immune system engage in harmonious interactions. Despite the significance of these relationships for host health, the dynamic immune operations that sustain mutualistic interactions at intestinal mucosal sites were long studied only by a small cadre of researchers, and this “frontier of the immune system” [6, 7] long remained the “poor relation of the immunology world” [8]. A rapid increase in our understanding of the composition and dynamic operations of the microbiome [9–12], coupled with striking examples of how the microbiome shaped intestinal immunity [13–18], kindled interest in identifying the most potently immunomodulatory microbiome members, and defining the mechanisms that underlie these interactions.
The inflammatory potential of the gut microbiome necessitates adaptations that prevent unfettered access to host tissues and their embedded immune systems, as well as immunologic adaptations that prevent deleterious responses following exposure to microbiome-derived products from beneficial or harmless microbes. The intestinal epithelium and its overlying mucus layer provide an effective physical barrier that limits exposure of the underlying intestinal immune system to the products from the gut microbiome. The critical nature of this physical barrier is exemplified by the deleterious impacts of impaired epithelial barrier function on host health. Despite its effectiveness, immune cells in the intestine, mesenteric lymph nodes (MLN), and gut-associated lymphoid tissues (GALT) are continuously being exposed to immune stimulatory products and foreign antigens (defined here as antigens not encoded in the host genome) [19, 20]. Upon encounter with these factors, the intestinal immune system must respond in a manner appropriate to the nature of the challenge. As such, the immune system functions as a sophisticated decision-making device that integrates quantitative and qualitative features of its environment to mount a response that is tailored to effectively manage the various entities it encounters. Making the “correct” decision is critical as inappropriate inflammatory responses to harmless/beneficial microbiome members promote tissue destruction, while tolerogenic responses that limit the capacity to restrain harmful pathogens can lead to severe illness or death. Despite these challenges, the immune system reliably generates appropriate responses, effectively balancing protective immunity against pathogens while maintaining mutualistic interactions with the microbiome.
How the immune system achieves this balance has intrigued immunologists for the past few decades and prompted efforts to define the molecular and cellular mechanisms through which microbiome members elicit tolerogenic host immune responses despite exposure to immune-stimulatory compounds and non-self-antigens from both symbiotic and pathogenic bacteria. Ultimately, intestinal immune tolerance is shaped by the cooperative function of both immune and non-immune cells in response to microbial signals derived from the microbiome. Central in this intricate network are CD4+ T cells, which facilitate the coordination of finely tuned and highly microbe-specific responses to neutralize threats or maintain intestinal homeostasis. Although the study of immune tolerance in the intestine has a rich history built on the investigation of tolerance to orally administered antigens [8, 21], the capacity of gut microbes to induce immune tolerance has received considerably less attention until recently. Here, we review the current understanding of how intestinal CD4+ T cell responses to the gut microbiome are generated, tracing the immune response from the initial acquisition of antigen to the final accumulation of polarized effector CD4+ T cells in the gut, and highlight the many gaps which still remain in our understanding of the intricate networks involved. Many outstanding studies regarding the impact of the microbiome on other key cell types, body sites, and temporal aspects of these responses are not discussed here due to space constraints, and we recommend to the reader several outstanding reviews that address these interactions [22–25], thus providing a comprehensive understanding of immune microbiome relationships.
CD4+ T cell responses to the microbiome
CD4+ T cell activation is driven by recognition of antigens presented on major histocompatibility complex (MHC) II by antigen presenting cells (APC), where they can be recognized by the T cell receptor, in conjunction with costimulatory signals from the APC [26]. One of the central features of CD4+ T cells is their capacity to adopt a specific cellular fate depending on the nature of the stimulus to which it reacts. As such, CD4+ T cells can adopt a variety of fates, including T helper (Th)1, Th2, Th17, Th9, regulatory T cell (Treg), and T follicular helper (Tfh) that are characterized by expression of particular transcription factors required to program stereotypical patterns of cytokine secretion. Although it is clear that these fates exist on a continuum [27–29], this framework has proved a useful means to conceptualize how CD4+ T cell function is shaped in response to microorganisms [26]. Unsurprisingly, there exists a close relationship between the gut microbiome and CD4+ T cell phenotype and function in the intestine [30]. Notably, variation in microbiome composition contributes to variance in intestinal T cell phenotypes, and as such, microbiome composition represents a driver of inter-individual intestinal CD4+ T cell phenotypes [30]. Mounting an appropriate response to the microbiome is essential, as dysfunctional immune-microbiome interactions can precipitate autoimmune and inflammatory disorders. Thus, defining the molecular pathways that govern T cell responses to the microbiome is critical for understanding intestinal immune tolerance, and offers an opportunity to derive insights into how the immune system can be programed to promote tolerance to foreign entities.
At steady state, the intestinal lamina propria CD4+ T cell compartment is dominated by FoxP3+ Tregs [13, 14, 31–33] and Retinoic Acid Receptor–related Orphan Receptor γt (RORγt+) Th17 cells [13, 15–17, 34] which accumulate in a microbiome-dependent manner. Although the anti-inflammatory functions of Tregs are fundamental to our symbiotic partnerships [35], it is increasingly evident that microbiome-induced Th17 cells can contribute to the quiescent immune-microbiome state through their anti-inflammatory [36] and anti-microbial functions [37], their inflammatory potential notwithstanding [38, 39]. Although tolerance was originally described as “a state of indifference or non-reactivity toward a substance that would normally be expected to excite an immunological response” [40], it is now more commonly conceptualized to include the role played by immune cells, such as Tregs, in actively maintaining a non-inflammatory state through potent anti-inflammatory functions in response to “substances.” Here, we will consider the role played by Th17 and Tfh cells during steady state conditions in promoting immune ignorance through anti-microbial function and antibody induction, respectively, in keeping with the originally envisioned meaning of the term tolerance, and representing a critical arm that ensures the maintenance of mutualistic responses. However, the potent inflammatory potential of intestinal Th17 cells [38, 39], which distinguishes them from Tregs whose “raison d'être” is to limit inflammatory responses, is critical when considering their role as mediators of “tolerance.”
The microbiome-dependent accumulation of intestinal Treg and Th17 cells prompted intense investigation to identify the microbiome members responsible. Intestinal Tregs are potently immunosuppressive and express a suite of anti-inflammatory molecules, including interleukin-10 (IL-10), that prevent inflammatory responses directed against the microbiome and host tissue [35]. Treg induction has been shown to be a property of several different bacteria including a consortia of Clostridia strains [13, 14], the Altered Schaedler Flora (ASF) [31], several members of the genus Bacteroides [32, 33, 41, 42], various Helicobacter species [43–46], as well as Bifidobacteria [47]. Additionally, FoxP3- IL-10-producing T regulatory 1 (Tr1) cells can be induced by Bifidobacterium breve [48] (Fig. 1). While the intestine is home to a population of Tregs that develop in the thymus (thymic Treg; tTreg) and can be found in germ-free mice, a large proportion of colonic Tregs are microbiome-elicited cells that are posited to arise in the periphery from naïve precursors (peripheral Treg; pTreg), although formal demonstration of peripheral differentiation has only been obtained for a handful of microbes. Although no perfect marker for pTregs exists, colonic Tregs are enriched for cells that express the transcription factor RORγt in conjunction with FoxP3 [33, 49, 50]. Prior to the identification of RORγt as a pTreg marker, earlier studies utilized expression of HELIOS [51] and Neuropilin-1 [52, 53], as low or no expression of these markers had been linked to pTreg development using transgenic TCR expressing T cells and these cells were largely absent from germ-free mice. However, the use of HELIOS and Neuropilin-1 as definitive markers of pTregs has since been challenged [54–56]. While cell transfer studies support the concept of RORγt marking cells that arise from naïve precursors in the periphery, important limitations also exist [57–59], and it seems likely that no marker that can truly differentiate between pTreg and tTreg exists. RORγt expression in intestinal Tregs is microbiome-dependent, and microbiome-specific intestinal Tregs are typically uniformly RORγt+; however, it remains to be addressed whether all RORγt+ Tregs are microbiome-reactive. Moreover, as RORγt-FoxP3+ cells can proceed to being RORγt+FoxP3+ [60], it is possible that self-specific cells that develop intra-thymically as RORγt-FoxP3+ may also form part of the RORγt+ Treg pool in response to microbial stimulation, confounding the utility of RORγt as a marker of pTreg. pTregs play a critical role in limiting intestinal inflammation, with deletion of RORγt within the FoxP3+ compartment leading to development of more severe intestinal inflammation [33, 50, 61], likely attributable to declining FoxP3 expression in RORγt-deficient Tregs [61]. While these studies suggest that pTregs play a critical role in limiting pro-inflammatory responses in the gut, it is unclear if this is due to the loss of Tregs specific for gut microbiome members or some facet of RORγt function itself that drives tolerogenic programs in response to the microbiome. These findings have been interpreted to demonstrate a unique requirement for RORγt+ Treg at mucosal surfaces, however, it is likely that intestinal homeostasis is achieved by the dual activity of tTregs and pTregs as has been demonstrated for the prevention of colitis in the T cell transfer model [62]. To date, a lack of tools for the specific depletion of tTregs has limited further insights into their contribution to intestinal homeostasis.
Schematic of mechanisms of bacterial colonization, antigen acquisition, and presentation to CD4+ T cells in the intestine. Created in BioRender. Engelhart, M. (2025) https://BioRender.com/s6vv57i.
The induction of Th17 responses is somewhat more selective and was initially thought to be unique to segmented filamentous bacteria (SFB) colonization [15–17]. Now, a range of microbes, as well as the parasite Tritrichomonas musculis, have been identified to have Th17 inducing capacity [13, 34, 63]. While Th17 cells are understood to develop in the periphery, the microbiome can impact the thymic selection of SFB-specific αβ T cells [64]. Interestingly, microbiome-elicited Th17 cells are typically more dominant in the small intestinal lamina propria [16, 17, 34], while microbiome-elicited Tregs accumulate in the colon [13, 14, 16, 17, 31–34]. It is unknown whether site specific T cell polarization is a feature of microbe localization or a form of tissue control [67] whereby the fate of microbiome-reactive cells is imprinted by local tissue factors such that an optimal response for that specific tissue is generated.
Despite the pathogenic potential of Th17 cells in other tissues, microbiome-elicited Th17 cells are typically not inflammatory and benefit the host through their impact on microbiome composition [37], improvement of intestinal barrier function [37], and providing pre-existing immunity to combat pathogens [16]. For example, SFB-induced intestinal Th17 cells contribute to intestinal homeostasis by limiting the expansion of SFB itself, which can elicit pathogenic responses when its abundance is not contained [37. Furthermore, SFB induces a self-restraining anti-inflammatory feedback loop that includes the production of IL-10, that limits the pathogenic function of SFB-induced Th17 cells [36]. Both Tregs and Th17 cells further contribute to immune homeostasis through expression of a tissue reparative program characterized by expression of amphiregulin that likely helps to ensure tissue integrity, as at other body sites [68, 69], although amphiregulin has been linked to the development of fibrosis [70, 71]. The transcription factor c-MAF is critical for the development of these programs in both Tregs [45] and Th17 cells [36] and the collective actions of these cells serves to dampen inflammation through distinct and overlapping mechanisms to maintain immune-microbiome mutualism.
Besides Tregs and Th17 cells, the promotion of intestinal Tfh responses by the microbiome has been recently uncovered and appears to represent a cell fate induced by many gut bacteria [45, 72]. Interestingly, when Akkermansia muciniphila is colonized in mice together with the ASF, a Tfh program is the dominant fate adopted by A. muciniphila-specific CD4+ T cells, but in the context of a more complex microbiome, A. muciniphila-specific Treg, Th17 and Th1 cells emerge in addition to Tfh cells, and thus microbiome variation may shape the output of CD4+ T cells in trans [22]. Although the role of these microbiome-specific Tfh cells has not been extensively explored, it is likely that they promote the generation of high-affinity immunoglobulin (Ig) A antibodies that target the microbe for which these T cells are specific. Intestinal IgA plays a fundamental role in promoting host-microbiome mutualism [23, 73, 74] and is thought to preferentially coat immunogenic microbes to prevent penetration into host tissues and neutralize microbially derived inflammatory products [75, 76]. Microbiome-specific T-independent IgA exists [77], suggesting that intestinal T cells are not required for the generation of microbiome-specific IgA but instead form a feedback loop whereby IgA is induced by select microbes to self-regulate gut microbiome composition [78, 79]. Thus, microbiome-driven modulation of intestinal T cell fate can contribute to tolerance through modulation of IgA responses. Strikingly, although Tregs [80] and Th17 cells [81] have also been linked to induction of class switching to IgA, several reports show that Th17 cells and Tregs can adopt a Tfh fate as a means to shape intestinal antibody production [82, 83]. Although a more comprehensive review of the impact of the microbiome on intestinal antibodies is beyond the scope of this review, this topic has been extensively reviewed elsewhere [23, 73, 84].
In summary, the collective activity of intestinal CD4+ T cells sustains mutualistic interactions with the gut microbiome. This can be mediated through active inhibition of inflammatory pathways by Treg cells, through their production of cytokines like IL-10, but additionally through Th17 cells which promote immune ignorance by limiting the exposure if the host to various microbial products, both through promoting intestinal barrier function, anti-microbial function, as well as limiting host penetration by the microbiome through the generation of microbiome-specific antibodies. However, it remains to be fully understood how all of these functions are coordinated.
First encounters
The first step in priming CD4+ T cell responses involves the acquisition of antigen that must be presented to naïve CD4+ T cells by antigen-presenting cells (APC). Orally administered model antigens have been extensively used to define the uptake of antigen in the intestine [85]. However, we still have a limited understanding of the form that microbial-derived antigens are first encountered and if the same pathways mediate microbial and dietary antigen uptake.
Several means of antigen acquisition from the intestine have been uncovered using orally delivered free antigen. Microfold (M) cells which overlie the follicular associated epithelium (FAE) of GALT represent one of the best described processes. M cells are largely devoid of the thick overlying mucus layer that characterizes the intestine, which is believed to allow them to be efficient points of antigen acquisition and both dendritic cells (DC) and B cells (through a DC-independent process) can capture antigen from/through M cells [86–88. The availability of M cell deficient mice has allowed assessment of the requirement for these cells in antigen uptake, and revealed a role for M cells in the generation of Tfh cells and subsequent IgA in the Peyer’s patches (PP) [89]. However, despite their clear role in mounting immunity to pathogens [85, 90], there is a paucity of information regarding their role in uptake of specific microbiome members, or if their capacity to respond differently to distinct bacteria [91] plays a role in conditioning the intestinal T cell response. M cells are prevalent in the PP of the small intestine, however, the colon is the site of the densest colonization by the microbiome but lacks PPs. Instead, the colon harbors a caecal patch and colonic patch which represent induction sites for B and T cell responses [92, 93]. Moreover, solitary isolated lymphoid tissues (SILTs), including both cryptopatches (CP) and isolated lymphoid follicles (ILF), are distributed along the length of the small intestine and colon in both mice and humans, representing alternative induction sites to the PPs [94, 95]. Given that they also possess M cells [96, 97], ILFs are likely an important site of induction of adaptive immune responses in the colon, functioning akin to PPs [97, 98]. Moreover, ILFs are microbiome-responsive [99], and bacterial species such as SFB can induce the development of secondary lymphoid follicles that act as induction sites independently of PPs [100]. Extensive variation between individual ILFs in the small intestine suggests they likely function as unique inductive sites, tailoring to the needs of individual regions of the intestine [101]. To date little is known about the requirement for these structures in the coordination of responses to the microbiome, but the distinct development requirements in mice between small intestine and colon ILFs may afford the opportunity to tease apart their role in mediating responses to the microbiome [102, 103]. Difficulties with the removal of ILF structures prior to phenotyping of intestinal tissue, allied to challenges associated with the genetic depletion of ILFs, has made it challenging to uncover their contributions to intestinal T cell function [104, 105]. However, elegant studies in humans where these structures can be visualized and removed have led to the identification of two types of ILFs, mucosal-ILF and submucosal-ILF, that differ in their location within the tissue and their cellular composition [105]. Thus, ILFs located at different sites along and within the intestine likely provide unique functions. While the presence of naïve T cells in these structures suggests that they may represent a site at which microbiome-specific cells are primed [105, 106], further studies are required to delineate the contributions of these distinct sites to the polarization of microbiome-specific T cells.
As an alternative to M cell uptake in GALT, it has been posited that some APCs acquire luminal antigen by extending long cellular protrusions, or transepithelial dendrites (TED), through the epithelial layer [107–110]. While this process has been visualized via two-photon microscopy, the lack of tools that would allow for deletion of this function has meant that it is difficult to assess the relative contribution of this process to antigen acquisition. More recently, mucus-secreting goblet cells have been demonstrated to possess antigen uptake capabilities [111, 112]. These cells readily take up antigen and particles through goblet cell-associated antigen passages (GAP) via endocytosis and deliver the antigen to APCs via transcytosis [113]. Depletion of goblet cells using Math1^fl/fl^ X Vil^Cre-ERT2^ mice or inhibition of GAP formation through provision of luminal murine epidermal growth factor which inhibits GAP formation through the epidermal growth factor receptor (EGFR), reveals that GAP structures and goblet cells are required for the transfer of orally provided ovalbumin to APCs, and the optimal induction of anti-inflammatory pTregs in the small intestine [111, 114]. During intestinal Salmonella infection, GAP formation is inhibited, limiting the proliferation of CD4+ T cells specific for dietary antigen. This inhibition of GAP formation likely sustains tolerance to dietary antigens during infection by limiting their uptake together with pathogens that concurrently are driving a proinflammatory response [115]. Despite their prominent role in mediating immunity to dietary antigen, mice with impaired GAPs still harbor robust populations of RORγt+ Tregs in the colon [111] suggesting that they are not required for induction of microbiome-specific Tregs at this site. Indeed, MyD88-dependent goblet cell-sensing of microbes actively limits colonic GAP formation to prevent inordinate inflammatory responses to the microbiome [114]. Therefore, it is possible that both the site of uptake and the source of the antigen may impact the route(s) of antigen acquisition, with the induction of tolerance to dietary antigens operating through distinct pathways to that of microbially-derived antigens. Much work remains to be done to track the pathways and forms through which microbiome-derived antigens are transferred to APC in the process of initiating microbiome-specific CD4+ T cell responses.
A growing body of work suggests that outer membrane vesicles (OMV) from Gram negative bacteria [116] and membrane vesicles (MV) from Gram positive bacteria and fungi [117] harbor immunomodulatory potential [118–123], and represent a strategy that allows the microbiome to intentionally shape intestinal immune responses. These vesicles can cross the epithelium [118, 119] and carry antigens that are recognized by microbiome-specific CD4+ T cells [42, 124], suggesting that the antigen acquisition by APC is through uptake of such vesicles rather than free antigen. Cell wall products can also cross the epithelium in vesicles or in vesicle-free form [119, 125] and may provide a source of factors that condition the response through stimulation of pattern recognition receptors (PRRs). Moreover, live bacteria can translocate to the MLN, potentially within live DCs, under homeostatic conditions [113, 126]. However, a dearth of tools that can precisely inhibit any of these processes within the bacteria themselves has limited our ability to determine which of these processes is most important for the stimulation of CD4+ T cell responses. Recent advances identifying signals that govern the localization of factors to such vesicles may allow assessment of this question.
The localization of microbes within the gut lumen or the mucosal interface has long been posited as an important factor governing their immunomodulatory potential. SFB, which represents one of the most immunogenic gut microbes identified to date [15, 16, 18, 127, 128], penetrates the intestinal mucosa and forms close associations with the host epithelium [129–131], possibly through the abundant glycosyl hydrolases and glycan utilization genes in its genome [132]. SFB adheres to intestinal barrier cells via use of a holdfast structure that attaches to the cell plasma membrane without rupturing the cell [124, 133, 134], and this attachment is thought to be critical for its immunomodulatory potential. Specifically, mouse-derived SFB potently induces Th17 cells in mice but not in rats, while rat-derived SFB potently induces Th17 cells in rats but not in mice [135]. Although both isolates show efficient cross-species colonization, mouse-derived SFB does not adhere to rat epithelium and vice versa [135, 136], thus supporting the concept that SFB induces Th17 accumulation only in hosts in which it adheres to the epithelium. Interestingly, SFB-specific Th17 cells are most enriched in the ileum, the site with most abundant SFB adherence, further corroborating the role of adherence in this response, possibly though increasing antigen availability [16, 135, 137]. SFB directly transmits vesicles containing CD4+ T cell antigens to the host via adherence-triggered endocytosis, and interference with these endocytic events through deletion of epithelial cell CDC42 impairs the ability of SFB to induce Th17 cells, despite normal attachment to intestinal epithelial cells (IEC) [124, 129]. Thus, Th17 induction is not mediated by epithelial adherence alone, but by the downstream transfer of SFB-derived material across the epithelium. While we lack similar levels of mechanistic insight for other microbiome members, Clostridia spp. [13, 14, 128, 138], Helicobacter spp. [44–46], Bacteroides fragilis [41], and A. muciniphila [72] are all potent modulators of intestinal T cell fate in steady state conditions and can reside in close contact with the host by colonizing the colonic crypts or the mucosa, suggesting that intimate contact with host epithelial cells at this location is important for immunomodulatory capacity. Although obtaining formal proof of this concept has been challenging, deletion of a BF3134-encoded glycosyl hydrolase in B. fragilis impairs mucus colonization, leading to a reduced induction of IL-10-producing Tregs [139, further suggesting that intimate proximity and contact with host epithelial cells is important for immunomodulatory capacity [72, 140–144].
Despite progress in our understanding of antigen acquisition by the intestinal immune system, the nature of the initial uptake of microbial antigens in the gut remains one of the most poorly understood aspects of immune interaction with the microbiome. Consequently, we have little information as to whether the mode of acquisition impacts the fate of CD4+ T cells or simply represents a highly redundant step to pass antigens to APCs for the activation and differentiation of intestinal T cells.
Unexpected antigen-presenting cell complexity
Following the acquisition of antigen, the next step in priming of CD4+ T cell responses to the microbiome requires antigen presentation by a “professional” APC that can activate a naïve CD4+ T cell. APCs are critical regulators of CD4+ T cell fate following activation, sensing conserved microbial products through PRRs in order to provide distinct T cell polarizing signals that elicit the differentiation of the appropriate cell subset [26]. APCs thereby link microbial features with specific antigens to help ensure a response that both coordinates appropriate downstream immune functions and is specific to the eliciting agent. The superior capacity of conventional/classical dendritic cells (cDC) relative to other APCs to migrate from sites of antigen uptake to draining lymph nodes where a large pool of naïve CD4+ T cells are available for activation, identified them as the most likely cell type for priming microbiome-specific T cell responses [145–147]. Intestinal cDCs are CD11c+MHCII+CD64- cells dominated by three predominant subsets: CD103+CD11b-(cDC1), CD103+CD11b+ (cDC2), and CD103-CD11b+ (cDC2) [148, 149]. Elegant ex vivo assays demonstrate that these DCs are the predominant capturer of intestinal antigen for priming in the MLN, endowed with the capacity to migrate to the MLN and promote Treg [150, 151] and Th17 [152] differentiation under steady state conditions. Various genetic systems that facilitate the depletion of individual cDC subsets further suggested a distinct role for different subsets of DCs in the priming of Treg or Th17 responses [153–156]. However, more recent efforts using newer tools to delineate the precise antigen presenting cells that promote the development of microbiome-specific T cells has uncovered previously unappreciated complexity.
Examination of the APC requirements for the induction of SFB-specific Th17 responses revealed that MHCII expression by CD11c+ cells is both essential and sufficient [66], as would be expected for a cDC-driven event, and was consistent with the reduced intestinal Th17 accumulation reported in mice lacking CD103+CD11b+cDC2 [153–156]. However, this was subsequently posited to be due to CD11c+ macrophages as “FMS-like tyrosine kinase 3 ligand” (Flt3l)^-/-^ mice that have severely reduced cDCs [157] showed unimpaired generation of SFB-driven Th17 cells [158]. Furthermore, C-C chemokine receptor 2 (CCR2)-mediated depletion of macrophages, or anti-colony stimulating factor (CSF)1-mediated inhibition of macrophage development, significantly reduced the accumulation of SFB-driven Th17 cells, while monocyte transfer corrected this defect, thus collectively demonstrating a requirement for macrophages in the response. Importantly, SFB-induced Th17 induction can occur in the absence of all known secondary lymphoid tissues, including MLNs, in lymphotoxin A (LTα)-deficient mice [65, 66, 159], thereby providing a plausible mechanism by which macrophages, which do not migrate to the MLNs [160], could mediate SFB-specific Th17 cell induction. This is in contrast to other studies which suggest that MLNs are the site of SFB-specific Th17 cell induction under normal conditions [161, 162]. Furthermore, a separate study that used a combination of C-C chemokine receptor 7 (CCR7)-deficiency and depletion of cDCs using a ZBTB46-diphtheria toxin receptor (DTR) system where diphtheria toxin (DT) administration ablates cDCs [163], suggested that cDC migration to the MLN is required for SFB-specific Th17 cell induction [162]. While the reason for this discrepancy is unclear, it is known that although substantially impaired, Flt3l^-/-^ mice develop a limited number of cDCs [157], and it is possible that these cells are absent in ZBTB46-DTR mice following DT treatment. Furthermore, while cell depletion strategies can uncover a role for a specific cell type, they do not establish them as the critical cell for presentation of antigen. As macrophages can transfer orally-fed antigen to CD103+ DCs via connexin 43 [164], the critical role for macrophages in priming of SFB-specific responses may be in the capture and transfer of SFB antigen to the migratory DCs that activate naïve T cells, but it is as yet unknown if such transfer occurs with microbiome-derived antigen. Finally, the studies showing a role for macrophages assessed Th17 cell development in response to SFB colonization in a polyclonal CD4+ T cell repertoire, while those showing a role for cDCs assessed Th17 development in a monoclonal TCR transgenic system with specificity for a single SFB-encoded antigen. Thus, it is possible that differences in the approaches used further complicate final interpretation.
Studies have also proposed that cDCs prime responses to other gut microbiome members, as cDC ablation using the ZBTB46-DTR system leads to a failure to accumulate Helicobacter-specific CD4+ T cells in the MLN [43]. Despite proposals that CD103+ DCs are the critical drivers of Treg development [150, 151, 156], none of the CD103+ subsets previously established to have potent Treg-inducing capacity are required for Treg generation in this system [165]. While the residual development of CD103+ DC subsets in the various genetic-depletion systems used means that a requirement for these cells cannot be unequivocally ruled out, these data suggested a highly redundant process whereby any of a variety of cDC subsets could support the development of Helicobacter-specific Tregs. Notably, prior work with the same Helicobacter-specific T cells using an alternative mechanism of depletion of CD103+CD11b+ cells via CD11c-Cre driven deletion of Notch2, did impair Treg development [43]. This discrepancy highlights how the specific depletion systems used may impact interpretation rather than simply the loss of a particular cDC subset. In a separate study of H. hepaticus-specific CD4+ T cells, in keeping with the concept that the MLN is the site of Treg induction in H. hepaticus-specific CD4+ T cells [166], CCR7-dependent migratory CD11c+MHCII+ APCs were established as the required carrier of H. hepaticus antigens and the most potent activator of the canonical RORγt+ Treg program, consistent with a role for cDCs in this process [167]. Intriguingly, this induction of H. hepaticus-specific Tregs also required expression of the transcription factor RORγt by CD11c+ cells. As RORγt is uniformly expressed by type 3 innate lymphoid cells (ILC3) which had also been established to have antigen presenting capacity [168], and were concurrently proposed to prime H. hepaticus-specific Tregs [169], this called into question the identity of these cell types. Are they CD11c+ ILC3s, or cDCs that express, or formerly expressed, RORγt, or are they the newly described Thetis Cells (TC) [170], or autoimmune regulator (AIRE)+ Janus Cells (JC) [171, 172]?
In support of a DC subset driving pTregs, recent studies have described PRDM16-expressing APCs that are distinct from ILC3s, and whose development is dependent on a cis-regulatory element 7 kb upstream from the RORγt transcription start site, as the cell type that is essential for microbiome-driven and oral antigen-driven pTreg development [173, 174]. These cells transcriptionally resemble DCs rather than ILCs and have consequently been termed tolerizing DCs (tolDCs) [173]. In keeping with the primacy of these cells for pTreg induction, their numbers remain stable through at least 12 weeks of age [173] and could explain the generation of pTregs in mice long after the weaning period [167]. By contrast, significant evidence also supports TCs as the key pTreg generating cell type [170]. MHCII deletion in cDC1s and cDC2s using Clec9a-Cre [175] or in ILC3s using RORα-Cre does not impact accumulation of colonic pTregs, suggesting that neither cell type is responsible for pTreg generation [170]. Furthermore, TCs express CD11c, ZBTB46, RORγt, and CCR7 and have variable expression of prdm16 [170, 173, 176, 177], all of which have previously been used to identify cDCs or ILC3s as the cell type inducing microbiome or dietary antigen specific pTregs. Thus, it is likely that tools that deplete cells or delete genes using these markers also impact TCs. Interestingly, Helicobacter-specific Treg induction is superior in 3-week-old compared to 14-week-old mice [43], tracking with the developmental wave of TCs [170, 176]. However, despite the relative impairment of pTreg generation in older mice, even at 14 weeks old, a sizeable population of pTreg generations is produced [43], suggesting that either a distinct APC is responsible or that the smaller numbers of TCs are sufficient. Despite different approaches to identify tolDCs and TCs, these cells are highly similar and may represent overlapping cell types, and much work remains to clarify the relationship of these cells to cDCs. At present, such cells have yet to be purified in sufficient quantity to demonstrate this capacity in an ex vivo setting, likely due to the enormous technical challenges in doing so. Thus, while limiting MHCII expression to these cells appears to be sufficient for pTreg generation, it is not yet clear if this occurs in concert with other cell types for full maturation of the response. Although a role for AIRE + JCs in mediating pTreg development in response to the microbiome has not been established, the depletion of AIRE + APCs negates pTreg generation in response to oral antigen, although this does not require the activity of AIRE itself [171].
Despite many similarities between the induction of tolerance to orally-provided antigen and microbiome members, restricting MHCII expression to RORγt+ cells is sufficient to support robust H. hepaticus-specific RORγt+ pTreg development [167]. In contrast, RORγt+ cell expression of MHCII is only sufficient to induce FoxP3 but not RORγt expression in response to orally-provided antigen [178], and cDCs may additionally contribute to the full maturation of these cells. While mechanisms that mediate the tolerogenic effects of these cells are not fully defined, expression of components of the Transforming Growth Factor (TGF)-β activating αvβ8 integrin in CD11c+ cells (αv) [167] or RORγt+ cells (β8) [170] is critical for pTreg induction. Furthermore, using a mixed bone-marrow chimaera approach that allowed for the generation of mice where APCs could either present MHCII or activate TGF-β via αvβ8 integrin, but not both, it was shown that TGF-β activation and antigen presentation must be performed by the same cell type [170]. TGF-β has long been linked to the induction of Tregs from naïve precursors [179], however it has been challenging in vivo to distinguish between direct effects of TGF-β on Treg induction, from the pro-inflammatory environment induced following neutralization of the effects of TGF-β [180, 181]. The transfer of Helicobacter-specific CD4+ T cells expressing a dominant negative TGFβRII and thus having impaired responsiveness to TGF-β [180], into an otherwise normal host where all other cells received unimpaired levels of TGF-β stimulation, has suggested that the loss of TGF-β signaling in CD4+ T cells only has minor impacts on Treg differentiation [43]. The leakiness of the system used notwithstanding, these data suggest that rather than providing a signal for Treg development, the activation of TGF-β by TCs or PRDM16+ tolDCs might condition a tolerogenic phenotype in APCs in the face of stimulation with microbial products. This is evidenced by CD11c-Cre mediated deletion of the TGFβRI being linked with a reduced capacity to support pTreg development [182]. A prominent role has been shown for IL-10 in supporting development of microbiome-reactive Treg [44, 45], but it is not clear if this reflects a conditioning effect of IL-10 on the developing T cell, or the capacity of IL-10 to limit inflammatory responses in the face of microbial stimulation that would indirectly block Treg development. Notably, whether these novel APC possess the retinoic acid generating properties previously posited to drive Treg induction by intestinal DCs [150, 151] is unknown.
In the case of intestinal Th17 development, relatively little is known about the pathways inducing their development, however, for SFB-induced Th17 cells, IL-6 is required for early differentiation, but signals provided by other Signal Transducer and Activator of Transcription 3 (STAT3)-activating cytokines like IL-21 or IL-23 can support normative accumulation in the absence of IL-6 [161]. In addition, locally produced factors like serum amyloid A (SAA)1/2 are induced by SFB in epithelial cells and promote IL-17A expression in the lamina propria [137].
Despite these advances in our understanding of the APCs that induce tolerance, critical knowledge gaps still remain with respect to other cell fates. Although the role for TCs, and PRDM16+ tolDCs in priming H. hepaticus-specific pTreg is clear, H. hepaticus-specific FoxP3- T cells proliferate and acquire a Th17 cell phenotype in the absence of CCR7 or RORγt expression by CD11c+ or MHCII+ cells [167] or PRDM16-expression by RORγt+ cells [173]. This fact suggests that rather than a single APC type acquiring microbiome-derived antigen and polarizing the appropriate effector response based on features of the microbe, that instead, distinct APC types may acquire antigen and prime different responses to the same microbe. The identity of this APC that drives Th17 development in the absence of tolDC/TC is unknown, but both CD11c^low^CD70^high^ cells and migratory CD103-CD11b + DCs have previously been shown to induce Th17 cell responses and may represent a candidate APC type [152, 183]. Importantly, under homeostatic conditions, Th17 development among H. hepaticus-specific T cells is rare and is prevented by the CCR7+ APC-driven Treg development. However, whether CCR7+ APCs limit the acquisition of antigen by the Th17-priming subset under normal conditions, or whether their effect on CD4+ T cell fate is dominant, remains an open question. While the studies of SFB-driven Th17 cell development were largely performed prior to knowledge of TCs, tolDCs, or JCs, RORγt-driven deletion of MHCII enhances accumulation of Th17 cells in SFB-negative hosts but does not impact SFB-driven Th17 cell development [66]. Although originally thought to reflect the capacity of ILC3s to inhibit microbiome-reactive T cells [184], a failure to generate pTregs in these mice likely leads to enhanced differentiation and expansion of Th17 cells. Interestingly, while restriction of MHCII to CD11c+ cells supports robust differentiation of H. hepaticus-specific Tfh cells, restricting MHCII expression to RORγt+ cells does not. This suggests that the mixed CD4+ T cell fate that characterizes H. hepaticus-specific CD4+ T cells requires the function of distinct APCs. It is thus clear that there is unappreciated complexity that contributes to intestinal CD4+ T cell fate. Indeed, most work to date has focused on the cell type priming the initial response. However, it is possible that once activated, cells may modulate their output based on subsequent APCs they interact with, in addition to their location. SFB-specific cells acquire RORγt expression in MLNs, but maximal IL-17A secretion is triggered upon entry to the small intestine [137]. Similarly, *H. hepaticus-*specific CD4+ T cells adopt a Treg fate in the MLNs, but maximum effector function, including IL-10 expression, occurs in the lamina propria even when Treg development does not occur [166]. Thus, much work remains to be done to fully understand the complexity of APC antigen acquisition and presentation to CD4+ T cells in the context of microbiome-derived antigen.
Polarizing factors
Concurrently with stimulation of the CD4+ T cell through its TCR via antigens presented on MHCII, APCs integrate information about the nature of the agent whose antigens they are presenting to coordinate the development of T cells with an appropriate effector phenotype (Fig. 2). This qualitative assessment is largely mediated through the expression of PRRs which sense conserved microbial patterns that convey broad phylogenetic information such that the APC can then provide signals, often secreted cytokines, to appropriately tailor the T cell response that ensues to adequately deal with the agent of interest [26, 185].
T cell and APC interactions leading to polarization of effector CD4+ T cells. Created in BioRender. Engelhart, M. (2025) https://BioRender.com/arug0to.
The prototypic immunomodulatory microbiome-derived product is polysaccharide A (PSA), a capsular zwitterionic polysaccharide that is produced by B. fragilis [186, 187]. PSA is packaged into OMVs [119] and can induce the development of IL-10-secreting Tregs that limit Th17 cell development [125, 188, 189] in response to B. fragilis. The recognition of PSA is mediated through the dual engagement of Dectin-1 and Toll-like receptor (TLR)2 which recognize the polysaccharide component and covalently attached lipid moiety of PSA, respectively [41, 188, 190–192]. Strikingly, PSA is sufficient to mediate many of the beneficial effects of B. fragilis, and its expression is required for optimal induction of Tregs by B. fragilis [41, 189, 191]. More recently, cell surface polysaccharides from Bifidobacterium bifidum [47] and H. hepaticus [193] have been linked to the induction of anti-inflammatory responses in a TLR2-dependent but Dectin-1-independent manner [47, 193], while the presence of a capsule in Ruminococcus gnavus strains has been linked to enhanced anti-inflammatory intestinal responses [194]. B. bifidum induces the development of IL-10+ pTregs in the colonic lamina propria, mediated through cell surface β-glucan and galactan polysaccharides which induce a TLR2-dependent tolerogenic phenotype in DCs [47]. While the specific IL-10-inducing factor produced by H. hepaticus has not yet been identified, biochemical assays suggest it is carbohydrate in nature, and is also sensed through TLR2 [193]. Expression of IL-10 is critical to limit the development of pathogenic CD4+ T cell responses to H. hepaticus, suggesting that such carbohydrate factors are required to induce an anti-inflammatory response. While information regarding PRR agonists from other microbiome members that coordinate CD4+ T cell function is scant, the induction of colonic Tregs by ASF and IL-10-secreting Tr1 cells by Bifidobacterium breve rely on recognition by host-encoded PRR, being MyD88- and Ticam-dependent, and TLR2- and MyD88-dependent [31, 48], respectively. Outside of TLR2-mediated recognition, TLR9-mediated sensing of microbiome-derived DNA can inhibit Treg accumulation in the colon, and strikingly, select microbiome members such as Lactobacillus spp. can inhibit TLR9 recognition to facilitate Treg generation. By appropriately regulating the size of the Treg compartment through TLR9, distinct microbes can maintain resistance to pathogens by tuning the ratio of Tregs to effector T cells [195], while others can block TLR9 stimulation to sustain Tregs which is protective in the context of intestinal infection [196]. Much less is known about the nature of the microbial signals that promote Th17 differentiation, but intraperitoneal administration of SFB flagellin has been shown to induce Th17-related responses in intestinal tissues [197].
The paucity of cell wall products that have been biochemically characterized has limited our understanding as to whether intestinal homeostasis is mediated by an assortment of microbiome-derived TLR2 agonists or by a single universal agonist common to many microbes. Furthermore, although stimulation of TLR2 via microbiome-derived agonists plays a prominent role in inducing immune homeostasis through their impact on T cells, TLR2 also mediates resistance to intestinal pathogens [198], and thus sensing of TLR2 agonists alone is insufficient to explain the connection between TLR2 and immune tolerance. Thus, biochemical characterization of a greater number of microbiome-derived agonists is needed. However, the challenges associated with biochemical purification of individual components from whole cells have long confounded the identification of PRR agonists [199–201]. In the case of PSA, the TLR2-stimulatory capacity was long-attributed to the polysaccharide component until a previously unappreciated covalently attached lipid was identified as key to the IL-10 inducing capacity of PSA [190]. Coupled with the propensity of TLR2 to sense lipids, this suggests that perhaps the activity of additional cellular products that operate via TLR2 are dependent on the presence of lipids that may have been overlooked. Efforts to study the necessity of these factors through gene deletion have been limited without the genetic characterization and technical understanding to delete the genes that coordinate their production, many of which likely cannot be deleted as they encode products that are sensed specifically because they cannot be turned off which would allow for easy immune evasion. Although it has been posited that such molecules may represent inherently tolerance-inducing entities, PSA was originally described as a driver of abdominal abscesses [187], and thus it is likely that the nature of the encounter, not simply the biochemical features of such molecules, dictates the development of tolerance. Much work remains to be done to better understand how these factors shape immune responses to microbiome members and to determine if there is a common biochemical feature that truly favors, or is conducive to, induction of CD4+ T cell tolerance.
The second class of molecules that guide the coordination of helper T cell function upon activation are metabolites that are produced or modified by the microbiome (referred to simply as microbial/microbiome metabolites hereafter). Short-chain fatty acids (SCFAs) are among the most prominent mediators of microbiome impacts on the host. SCFAs are the product of microbial fermentation of fiber, resistant starches, and otherwise indigestible carbohydrates, and represent one of the canonical functions provided by the gut microbiome [202, 203]. Intestinal SCFAs are dominated by acetate, propionate and butyrate, and are almost entirely absent in germ-free mice [202]. SCFAs reach their highest concentrations in the colon and although they have now been linked to phenotypic imprinting of numerous cell types [203], one of their earliest ascribed roles in immunomodulation was that of Treg-induction [204–206]. While the impact of genetic ablation of SCFA-producing ability on immune function has not been reported to our knowledge, the exogenous administration of butyrate, propionate or acetate can all promote Treg accumulation in the colon or the MLN [204–206], although the precise site affected differs between studies. The specific SCFAs produced varies across different classes of bacteria with acetate produced by a wide range of microbes, while propionate and butyrate production are more specialized. This redundancy with respect to Treg generation likely ensures that Treg-inducing capacity can be maintained across distinct microbial community structures where the precise composition of the SCFA pool may vary [207, 208]. Mechanistically, SCFAs mediate their functions both through sensing by specific host-encoded receptors as well as through modification of cellular epigenetic state and epigenetic regulators. Propionate and butyrate both engage host-expressed receptors to drive Treg accumulation in the colon, with propionate acting through GPR43 [204] and butyrate through GPR109a [209] (also a receptor for niacin). However, following passive diffusion or Slc5a8-mediated uptake [210], both propionate and butyrate can also directly inhibit the function of histone deacetylases (HDAC) through chemical modification [211, 212]. Furthermore, butyrate can provide a source of acetyl-CoA that leads to enhanced histone acetylation [213]. These mechanisms have been implicated in the capacity of butyrate to enhance histone H3 acetylation at the FoxP3 promoter [206] and conserved noncoding sequence (CNS) 1 and CNS3 [205, 206], which support the peripheral induction and enhance the efficiency of FoxP3 expression respectively [214]. Although butyrate can directly promote FoxP3 expression in naïve T cells [205, 206], it can also modify DC function to promote Tregs through inhibition of HDACs [205]. Despite a variety of studies replicating these findings, other studies have found no role for SCFAs in mediating Treg induction [33]. Furthermore, how the effects of SCFAs are limited to induction of FoxP3 and not to other CD4+ T cell fate-specifying transcription factors [206] remains unclear, but suggest that additional unknown factors are involved.
Another class of molecules which affect host physiology and are modified by the gut microbiome are bile acids. Bile acids are a set of compounds generated by the host from cholesterol in the liver that are secreted into the intestine to aid in lipid absorption. These primary bile acids can be modified by members of the microbiome to generate secondary bile acids, which can in turn shape host physiology through recognition by host-encoded receptors. Most bile transformations are mediated by the actions of bile salt hydrolases (BSH), which are widely distributed among members of the microbiome, and 7-α-dehydroxylases, which are less prevalent [215, 216]. The effects of the microbiome on bile varies highly depending on the microbe performing the transformation. Furthermore, due to the recycling of secondary bile acids, multiple distinct microbes may modify a given bile molecule, and thus the pool of bile acids varies across individuals in a microbiome-dependent manner [215]. Given the broad impact of secondary bile acids on host physiology, their capacity to shape immune function has received considerable attention, although only a handful of microbially-modified bile acids have been shown to shape intestinal T cell responses. The exogenous administration of derivatives of lithocholic acid (LCA) is sufficient to both inhibit Th17 development and promote Treg accumulation. 3-oxoLCA and isoLCA, which are generated by the action of 3α- and 3β- hydroxysteroid dehydrogenase enzymes, antagonize the Th17-specifying transcription factor RORγt in CD4+ T cells to limit Th17 cell development [217, 218]. Another LCA derivative, isoalloLCA, actively promotes the expression of FoxP3 through the T cell-expressed nuclear receptor NR4A1 in a CNS3-dependent, but CNS1-independent, manner [217, 219], likely through enhancing the acetylation of the FoxP3 locus. Notably, the capacity of B. fragilis and Bacteroides thetaiotaomicron to modify bile acid pools via BSH but not 7-α-hydroxysteroid dehydrogenase, has been critically linked to the accumulation of colonic RORγt+ pTregs [220]. The generation of isoDCA by co-metabolism of bile by 7α-hydroxyl cleavage from cholic acid by Clostridium scindens and the 3α-hydroxyl epimerization activity of Ruminococcus gnavus hydroxysteroid dehydrogenases heterologously expressed in B. thetaiotaomicron also drives RORγt+ Treg accumulation [221]. As with SCFAs however, data showing a requirement rather than sufficiency for these molecules is currently lacking due to significant challenges associated with the genetic modification of many members of the microbiome. Collectively, these studies reinforce the notion that many secondary bile acids are indeed critical regulators of the CD4+ T cell phenotype in the intestine.
Other metabolites have also been linked to polarization of intestinal T cells fate, albeit much less is known about these molecules. Microbiome-derived adenosine triose phosphate (ATP) can stimulate CD11c^low^ CD70^high^ APCs to induce the accumulation of Th17 cells [183], although it is unclear given the ubiquity of ATP production if all microbes are capable of supporting Th17 cell development via ATP generation. Tryptophan availability has long been appreciated to represent a critical regulator of immunity. While its effects can be traced in part to sensing of its availability by the GCN2 kinase [222], the metabolism of tryptophan to generate products which agonize the aryl hydrocarbon receptor (AhR), also plays important roles in its effects on the immune system [223]. AhR is expressed in intestinal pTregs and controls accumulation of intestinal Tregs, suggesting that a microbiome-derived metabolite contributes to intestinal Tregs through AhR [224]. *Lactobacillus reuteri-*produced indole-3-lactic acid (ILA) induces the accumulation of CD4+CD8αα+ IEL in an AhR-dependent manner [225], while indole-3-propionic acid promotes mitochondrial respiration in intestinal CD4+ T cells in an AhR-independent manner, limiting their capacity to differentiate into Th1 or Th17 cells [226]. Finally, gut microbes have recently been shown to produce serotonin which can promote Treg generation [227]. Thus, the microbiome generates a broad array of secreted molecules that can shape CD4+ T cell phenotypes to promote immune tolerance.
The recognition of cell wall components is understood to allow direct linking of the antigen source to the specific PRR agonist. As these molecules are often physically linked and thus enter the same phagosome, this often links a particular effector response to select antigens [228]. Metabolites, on the other hand, represent secreted/released effectors, and therefore, their impact is likely not directly linked to T cells specific for the microbes producing the metabolites. Indeed, butyrate has been directly shown to support the development of Tregs to orally administered antigen [206]. Thus, metabolites likely broadly condition the intestinal environment to favour T cell responses that maintain tissue integrity and mutualistic interactions. As such, the sensing of these metabolites may act akin to effector-triggered immunity [229], whereby the presence of these metabolites indicates a healthy microbiome state to which the host immune system should be tolerant without direct sensing of all the community members. Furthermore, this would ensure tolerance toward myriad distinct microbiome compositions, so long as they provided benefits to the host in the absence of danger [230], and lack of these molecules may indicate pathogenic disruptions that necessitate a distinct CD4+ T cell response. Interestingly, although a Th1-promoting environment can drive microbiome-reactive CD4+ T cells toward a Th1 phenotype [231], SFB-reactive T cells maintain a Th17 bias when responding to SFB even in the face of a concurrent strongly pro-Th1 response [232]. Thus, the Th17-inducing signals of SFB are likely delivered together with its antigens and can either overcome pro-Th1 signals or occur in an anatomic location where such signals are not present. Understanding how these two distinct scenarios are controlled will be important for the delivery of tolerance-promoting therapies that by necessity will most often be administered in the context of ongoing inflammatory responses.
Antigen specificity
Irrespective of the specific APC presenting microbiome-derived antigen or the signals conditioning the outcome of the response, antigen-bearing APCs must ultimately encounter CD4+ T cells with a TCR specific for these antigens to activate and polarize a CD4+ T cell response. Deciphering whether the CD4+ T cell subsets that accumulate in response to the microbiome are specific for the inducing agent or whether they are comprised of cells with a range of specificities, including the inducing microbe and other antigens in the environment, is of fundamental importance. Two distinct, but not mutually exclusive, possibilities exist for microbiome-modulated CD4+ T cell differentiation. First, specific microbes may generate an environment that promotes particular effector types, even if the antigens are derived from microbes distinct from those conditioning the environment. The second possibility is that T cells of a particular effector class are specific only for the microbe that provides the polarizing signals. While the latter model has been thought to explain how the immune system avoids mounting inappropriate responses to distinct microbial entities, as discussed below, examples of environmental conditioning have been uncovered and are in keeping with the role of microbially produced or modified molecules like metabolites in driving CD4+ T cell fate.
Seminal studies on the emergence of microbiome-specific T cells came from interrogation of the ability of the pathobiont H. hepaticus to shape CD4+ T cell function. Using a combination of approaches, it was established that H. hepaticus elicited the development of H. hepaticus-specific Tregs with anti-colitic activity in the MLN [44]. Moreover, when key regulatory pathways were disabled in the context of a dysfunctional IL-10 axis, colitogenic H. hepaticus-specific CD4+ T cell populations expressing IL-17A and IFN-γ emerged that could drive intestinal pathology [233–236]. More recently, the development of TCR transgenic mice specific for gut microbiome encoded-antigens has reinforced these findings, revealing that CD4+ T cells with specificity for SFB [232], B. thetaiotaomicron [42], Helicobacter spp. (H. apodemus, H. hepaticus, and H. typhlonius) [43, 45, 46], and A. muciniphila [72] leads to the development of Th17 cells, Tregs, and/or Tfh in the intestine under homeostatic conditions. Moreover, ex vivo assays have further shown that a cocktail of Clostridia spp. [13] and B. bifidum [47] also induce the accumulation of Clostridia-specific or B. bifidum-specific Tregs, respectively. Collectively, these studies have provided unequivocal evidence that gut microbes can drive the differentiation of CD4+ T cells that are specific for antigens that they encode. However, what has remained less clear is whether these microbes create a milieu that favors the development of specific CD4+ T cell lineages in response to antigen exposure irrespective of the source of antigen, or if these microbes coordinate development of such cells specific solely for their own antigens.
While challenging to address, a limited number of studies support the notion that these microbes may promote an environment conducive to the polarization of desired effector classes. In the context of SFB-induced Th17 differentiation, ex vivo assessment of the specificity of Th17 cells using hybridoma based systems revealed that, although SFB-specific cells are highly abundant within the pool of intestinal Th17 cells, a significant number of Th17 cells specific for non-SFB antigens also develop [66, 232]. Although it is possible that these Th17 cells represented those that had differentiated prior to SFB colonization rather than being elicited by colonization with SFB, their preponderance in the Th17 pool (>50% of the tested fusions were not SFB-reactive in one study [66]) was disproportionate with the Th17 cell abundance in mice lacking SFB (Th17 abundance increased >5-fold following SFB colonization). The adoptive transfer of T cells specific for a non-SFB antigen has provided a more formal test of whether SFB induces these cells, but has also failed to provide a clear answer. In one such study, the adoptive transfer of CD4+ T cells from ovalbumin-reactive OT-II and melanocyte-reactive TRP-1 TCR transgenic mouse lines, Th17 development in the transgenic T cells was unaffected by SFB colonization, even with provision of their cognate antigen [66]. Conversely, using a similar experimental framework with OT-II TCR transgenic CD4+ T cells, a separate study found that SFB could enhance Th17 differentiation among transferred OT-II cells when ovalbumin was provided [65]. While this latter study did not use recombination activating gene (Rag)-/- OT-II, and thus could not completely exclude a role for additional specificities in the transferred cells due to secondary TCR rearrangement, stimulation with ovalbumin was required for Th17 development suggesting that triggering of a non-SFB TCR was critical for the response. Similarly, provision of the SCFA butyrate, promotes the development of ovalbumin-stimulated OT-II into Tregs in the colon [206], suggesting some microbiome members elicit an environment that is conducive to Treg development rather than simply inducing Treg development to their own antigens exclusively.
An additional layer of complexity in the study of antigen specificity is the inherent cross-reactivity of the TCR [237]. While the antigen-specificity of intestinal T cells has been largely studied in the context of individual microbes, it is likely that individual T cell clones harbor reactivity to other microbiome members due to shared epitopes and cross-reactivity. Although not extensively explored, several studies support a model whereby intestinal CD4+ T cell clones are reactive to multiple different species. This was first established for H. hepaticus using CD4+ T cell clones generated from H. hepaticus-colonized mice that are specific for the flagellar hook protein which were shown to be reactive to some, but not all, closely related Helicobacter species [238. A similar phenomenon was uncovered using a B. thetaiotaomicron-specific hybridoma generated from CD4+ T cells that are specific for an epitope derived from BT0900, but were also reactive to other Bacteroides species that had exact copies of this epitope [239]. More recently, a systematic study of the antigen specificity of intestinal CD4+ T cells expanded on these findings by identifying epitopes from a single antigen that were found in multiple distinct microbiome members and recognized by highly represented TCRs within the pool of intestinal CD4+ T cells [240]. Collectively, these data suggest that individual T cell clones can harbor specificity for several microbiome members, negating the need for a dedicated pool of cells with reactivity to individual strains. This points to a mechanism through which intestinal CD4+ T cells maintain broad reactivity to the enormous microbial complexity of the gut microbiome within the constraints of the available TCR repertoire and limitations on the size of the CD4+ T cell compartment.
Despite this evidence, it is unclear how the intestinal immune system could tailor the quality of responses appropriate for different microbiome members if there is wide-spread expression of the epitope for which the T cell is specific. In such a scenario, a Treg response targeted to one microbe may not be appropriate for a different microbe that shares the same epitope. If epitope sharing is most common among closely related species, the likelihood that deleterious agents gain the benefit of epitope sharing with beneficial microbes is limited. However, even closely related strains of B. fragilis can exert distinct impacts [41, 241] on the host depending on the presence of the B. fragilis toxin (BFT), suggesting that tuning the response at the level of individual isolates may be useful. Second, the mere presence of the DNA sequence that encodes the epitope in the genome does not guarantee recognition as B. thetaiotaomicron-specific T cells which recognize the antigen encoded by BT0900, fail to respond to B. caccae despite this bacterium sharing the identical epitope [239]. Similar findings have been reported for BθOM mice that express a B. thetaiotaomicron-specific MHC II-restricted TCR that responds to a BT4295-encoded epitope. In this model system it has been demonstrated that many, but not all, strains of B. thetaiotaomicron with the cognate epitope are recognized by the TCR [242].
To further complicate matters of antigen specificity, TCRs may also recognize both microbiome-encoded and self-antigens. Striking data have implicated a role for SFB as a driver of autoimmune pathology in extra-intestinal sites, including the pancreas [243], joints [244], and central nervous system [245, 246]. Although this could be mediated independently of SFB-specific Th17 cells, a recent study identified SFB-reactive Th17 cells that recognize self-antigens expressed in the central nervous system (the receptor tyrosine kinase ERBB2, trophinin 1, ANAPC2) that can induce central nervous system pathology [246]. A similar phenomenon has been demonstrated in a murine model of uveitis caused by CD4+ T cell reactivity to interphotoreceptor retinoid-binding protein (IRBP). IRBP-specific Th17 cells developed in the intestine in a microbiome-dependent, but IRBP-independent, manner, implicating cross-reactivity between IRBP and a microbiome-encoded antigen [247]. Notably, this was not thought to require SFB colonization. Little is known about the cross-reactivity between microbiome-reactive Treg TCRs and self-antigens, but it has been suggested that T cells with microbiome-reactive TCRs can acquire FoxP3 expression in the thymus [248]. This is in keeping with a study that identified tTreg development driven by a TCR with non-self-cognate specificity [249]. Whether the self-reactivity of microbiome-reactive TCR plays a role in their thymic selection or peripheral maintenance is unclear. However, at least in the case of SFB-reactive T cells, the affinity of the TCR for self-antigen is lower than for SFB-encoded antigens, and their pathogenic functions with respect to autoimmunity likely require priming by a microbial agent to generate the conditions to support their pathogenic function.
Conclusions
While study of the microbiome-driven differentiation of CD4+ T cell fate in the intestine has provided myriad insights into how homeostatic responses against non-genomically encoded antigens can be achieved, many questions remain to be answered to fully comprehend how the delicate balance of CD4+ T cell tolerance and effector functions is maintained. Our limited capacity to genetically modify microbiome members has contributed to our incomplete understanding of how microbiome members shape intestinal immune responses. Furthermore, despite the wealth of information that has been gathered to date, current knowledge is built off the study of a small pool of model organisms. While many of these findings may be broadly applicable to other bacterial species, a critical challenge of future work will lie in validating how universal these mechanisms of antigen acquisition, presentation, and induction of effector function are across the repertoire of microbes in our gut, and indeed, whether these mechanisms extend beyond the recognition of bacterial members of the microbiome. The renewed interest in oral tolerance [21] has revealed several overlapping features with induction of tolerance to the microbiome. Despite many similarities, differences in the requirements for tolerance to oral or microbial antigens exist [167, 178], and illumination of these differences will further advance our understanding of intestinal immune tolerance. Given the vast complexity of the microbiome, the task of unraveling these questions is undoubtably immense, but is of paramount importance in the continued effort to understand the relationship between the intestinal immune system and the microbiome. Moreover, as intestinal pTregs can limit responses to oral vaccines, the capacity to limit their development in select time windows may be of benefit [250]. The studies discussed here focused on work from murine models, which have provided the tools necessary to derive insights into the nature of CD4+ T cell-microbiome interactions. Emerging evidence suggests that these relationships are not unique to mice. Microbiome-specific CD4+ T cells are prevalent in healthy humans [251], and human-derived gut microbes coordinate the development of Treg [13, 32] and Th17 responses [13, 34] in mice, suggesting conservation of these responses between the two species. However, more studies are required to delineate the impact of specific gut microbiome members on intestinal T cell responses in humans. Considering the emerging evidence that the microbiome plays a pivotal role in myriad inflammatory diseases, both inside and outside of the intestine, understanding the relationship between intestinal T cells and the microbiome is critical to the development of durable therapeutic strategies that tailor interactions between host and microbe to promote/restore immune tolerance.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Owen RD . Immunogenetic consequences of vascular anastomoses between bovine twins. Science 1945, 102, 400–1.17755278 10.1126/science.102.2651.400 · doi ↗ · pubmed ↗
- 2Billingham RE, Lampkin GH, Medawar PB, Williams HL. Tolerance to homografts, twin diagnosis, and the freemartin condition in cattle. Heredity (Edinb) 1952, 6, 201–12.
- 3Burnet FM . The Production of Antibodies. New York: Macmillan, 1941.
- 4Burnet FM, Fenner F. The Production of Antibodies (2nd edn). In: Burnet FM, Fenner F (eds). Melbourne: Macmillan, 1949.
- 5Billingham RE, Brent L, Medawar PB. Actively acquired tolerance of foreign cells. Nature 1953, 172, 603–6.13099277 10.1038/172603 a 0 · doi ↗ · pubmed ↗
- 6Janeway CA Jr . Frontiers of the immune system. Nature 1988, 333, 804–6.2968519 10.1038/333804 a 0 · doi ↗ · pubmed ↗
- 7Blumberg RS, Mac Donald TT. Mucosal immunology: a frontier no longer. Mucosal Immunol 2008, 1, 3.
- 8Mowat AM . To respond or not to respond—a personal perspective of intestinal tolerance. Nat Rev Immunol 2018, 18, 405–15.29491358 10.1038/s 41577-018-0002-x · doi ↗ · pubmed ↗