Mutational landscape and risk estimates of DDR genes in Chinese ovarian cancer patients
Cuiyun Zhang, Bing Wei, Xia Xue, Qingxin Xia, Yi Wang, Lanwei Guo, Tingjie Wang, Li Wang, Junli Deng, Yuping Guan, Xiaoyan Wang, Lu Feng, Rui Wu, Ziqing Hu, Klaas Kok, Anke van den Berg, Yongjun Guo, Jun Li

TL;DR
This study identifies specific genes linked to ovarian cancer risk in Han Chinese women, offering insights for personalized risk management.
Contribution
First gene-specific ovarian cancer risk estimates for DDR genes in Han Chinese population.
Findings
P/LPVs in BRCA1/2 are strongly associated with high-grade serous carcinoma and family history.
RAD51D, RAD51C, and MSH2 show significantly elevated ovarian cancer risks in Han Chinese patients.
Population-specific risk estimates highlight the need for tailored screening and prevention strategies in China.
Abstract
Pathogenic/likely pathogenic variants (P/LPVs) in DNA damage response (DDR) genes are known ovarian cancer (OC) risk factors, but gene-specific risk estimates in Han Chinese remain unclear. To accurately assess the risk associated with DDR genes in the Han Chinese population to facilitate personalized risk management and enhance clinical decision-making. We performed next-generation sequencing of 45 DDR genes in 666 OC patients from Henan, China. Associations between P/LPVs and clinical features were assessed using chi-squared tests. Variant frequencies were compared with population controls (gnomAD and ChinaMAP databases) to estimate gene-specific odds ratios (ORs) using Fisher’s test. In Henan Ovarian Cancer patients, the median disease onset age was 53 years (range: 24–81), with 7.7% diagnosed before 40. Most patients had advanced disease (56.5% Stage III, 18.9% Stage IV), and…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
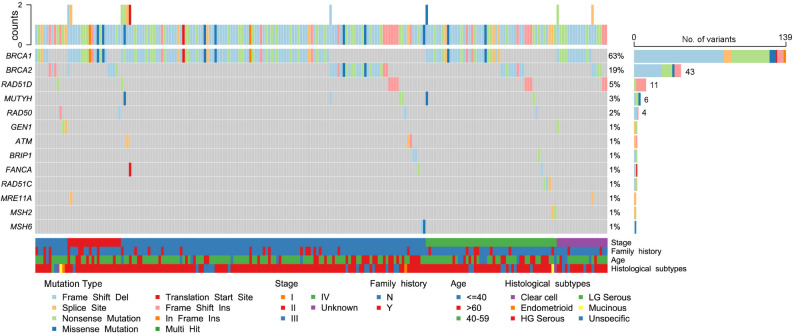 Figure 1
Figure 1- —https://doi.org/10.13039/100018925Health Commission of Henan Province
- —https://doi.org/10.13039/501100011447Science and Technology Department of Henan Province
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsBRCA gene mutations in cancer · Genetic factors in colorectal cancer · DNA Repair Mechanisms
Background
Ovarian cancer (OC) is the third most common gynecological malignancy characterized by a high mortality rate. The five-year survival rate for OC is highly stage-dependent, ranging from 15% in stage IV disease to nearly 95% in stage I. Unfortunately, over 70% of patients are diagnosed at advanced stages [1, 2]. In 2022, the estimated global incidence of OC was 324,398 cases, with 61,100 cases reported in China [3, 4]. Standard screening methods, including transvaginal ultrasound and CA-125 blood tests, have been shown to be ineffective in reducing the incidence of OC or improving survival outcomes [5], therefore, targeted management of high-risk individuals may provide a more efficient strategy to reduce both the incidence and mortality of OC.
Carriers of pathogenic/likely pathogenic variants (P/LPVs) in BRCA1 have a 20–30 times higher lifetime OC risk compared to the general population, while those with BRCA2 P/LPVs increased approximately 10–20 times [6, 7]. Other genes implicated with elevated OC risk include BRIP1, PALB2, RAD51C, RAD51D, and STK11, most of which are involved in DNA damage response (DDR) [8, 9]. Accurate risk assessment is crucial for managing the carriers of OC predisposition genes, particularly in facilitating informed risk-reducing bilateral salpingo-oophorectomy (RRSO) decisions that should balance between benefits and risks [10]. However, precise risk estimates of these genes to OC have not yet been determined in the Chinese population. This study aims to determine the precise risk estimates of DDR genes in Han Chinese to support tailored risk management strategies and improved clinical decision-making.
Methods
Study cohort
The patients enrolled in this study were admitted to Henan Cancer Hospital between April 2018 and December 2021 with a confirmed diagnosis of epithelial ovarian cancer by two independent pathologists. Those without testing for germline variants were excluded from this analysis. To minimize potential confounding from population stratification, we restricted our analysis to a highly homogeneous patient population, of whom 98.8% (658/666) self-identified as Han Chinese. For comparison, the China Metabolic Analytics Project (ChinaMAP) reference database, which includes approximately 85.4% (9,043/10,588) Han Chinese individuals, was used alongside the East Asian subset of the Genome Aggregation Database (gnomAD) to ensure close ancestral matching with our predominantly Han Chinese cohort. We did not perform principal component analysis (PCA) or adjust for genetic ancestry because individual-level genotypic and phenotypic data were not available for the gnomAD and ChinaMAP reference populations. This study was approved by the Ethics Committee of Henan Cancer Hospital (approval number: 2021-KY-0091-002), and written informed consent was obtained from all participants in accordance with the Declaration of Helsinki.
DNA extraction, targeted sequencing, variant calling, and classification
Genomic DNA was extracted from the leukocyte fraction of 500µL peripheral blood using the QIAamp Blood DNA isolation kit (Qiagen, Germany) to minimize circulating tumor DNA (ctDNA) contamination, then quantified by Qubit dsDNA HS assay (Life Technologies, USA). A total of 200ng DNA was subjected to next-generation sequencing library construction following optimized protocols as previously described [11]. Coding exons and flanking intronic regions (± 20 bp) of target genes were polymerase chain reaction (PCR)-amplified using the DDR 45-gene kit (Novogene Biotech, China). Investigated genes are listed in Table S1. Indexed samples were sequenced with 121 bp paired-end reads on a NextSeq 550 platform (Illumina, USA). Reads were aligned to the hg19 reference genome by Burrows-Wheeler Aligner (bwa-mem v0.7.17) [12]. BAM files were sorted, and duplicated reads were marked and removed using SAMtools (v1.9.0) [13], sambamba (v0.7.1) [14]. Germline variants were called by three independent algorithms: Genome Analysis Toolkit Haplotype joint caller (GATK-haplotype v4.1.7.0) [15], FreeBayes (v1.1.0.46) [16], and SAMtools (v1.9.0) [13]. Called variants were filtered using strict thresholds (minimum depth 100× and variant allele frequency ≥ 30%, variant quality ≥ 30), followed by annotation with snpEff software (v4.3.1t) [17]. Randomly selected P/LPVs were Sanger-validated using leukocyte-derived DNA. Variants were described according to the Human Genome Variation Society (HGVS) nomenclature and annotated using public databases, including ClinVar, gnomAD, dbSNP, and COSMIC, to help identify known germline variants and exclude likely somatic mutation hotspots. Subsequently, variants were classified by two geneticists independently using the five-tiered system proposed by the American College of Medical Genetics and Genomics (ACMG) and the Evidence-based Network for the Interpretation of Germline Mutant Alleles (ENIGMA).
Statistical analysis and risk estimates
Categorical variables, including disease onset age, disease stage, histological subtypes, and family history, were compared across BRCA1/2^+^ (with P/LPVs in BRCA1 or BRCA2 genes), DDR^+^ (with P/LPVs in non-BRCA1/2 DDR genes), and DDR^-^ (negative for any DDR P/LPVs) patients using the chi-squared test. Odds ratios (ORs) and 95% confidence intervals (CIs) for each gene were calculated by Fisher’s exact test. Risk estimates for each gene were determined by comparison with their prevalence in the East Asian population of gnomAD (v4.1.0, https://gnomad.broadinstitute.org) and ChinaMAP (v2020-03.beta, www.mbiobank.com) [18, 19]. To correct for multiple hypothesis testing while preserving statistical interpretability, Benjamini-Hochberg false discovery rate (FDR) correction was applied only to genes observed in ≥ 2 Henan ovarian cancer (HOC) patients with a nominal p-value < 0.1 in either comparison. All statistical analyses were conducted using R software (v4.3.1), and two-sided p-values < 0.05 were considered statistically significant.
Results
Clinical characteristics of the patients
The median age at disease onset among HOC patients was 53 years (range: 24–81). No significant differences in age at disease onset were observed across subgroups: BRCA1/2^+^ patients had a median onset age of 52 years (range: 33–80), DDR^+^ patients had a median age of 53 years (range: 33–75), and DDR^−^ patients had a median age of 54 years (range: 24–81). Early-onset disease (< 40 years) was observed in 7.7% (51/666) of patients, with no significant difference among the BRCA1/2^+^, DDR^+^, and DDR^−^ patients (Table 1). In terms of disease stages, 10.2% (68/666) of patients were diagnosed at stage I, 6.9% (46/666) at stage II, 56.5% (376/666) at stage III, 18.9% (126/666) at stage IV, and 7.5% (50/666) with unknown stage status. Chi-squared analysis revealed a significant difference in stage distribution between BRCA1/2^+^ and DDR^−^ patients (p = 0.0084), notably with stage I disease being less frequent in BRCA1/2^+^ patients, suggesting a potential gap in the implementation of targeted screening and risk management for BRCA1/2^+^ carriers in this cohort.
Table 1. Clinical characteristics of Henan Ovarian Cancer (HOC) patients included in this studyCharacteristicsTotal (n = 666)BRCA1/2^+^ (n = 181)DDR^+^ (n = 50)DDR^−^ (n = 444)p-valuecases%cases%cases%cases%BRCA1/2^+^vs. DDR^+^BRCA1/2^+^vs. DDR^−^DDR^+^ vs.DDR^−^<40 (years old)517.66%94.97%36.00%398.78%1.00.140.69≥40 (years old)61592.34%17295.03%4794.00%40591.22%Stage I6810.21%84.42%612.00%5412.16%0.0980.00840.22 II466.91%1910.50%24.00%265.86% III37656.46%10055.25%2346.00%25858.11% IV12618.92%3720.44%1530.00%7516.89% Unknown507.50%179.39%48.00%316.98%Histological Subtypes High Grade Serous50575.83%15686.19%3672.00%32172.30%3.26e-071.04e-050.38 Low Grade Serous203.00%10.55%00.00%194.28% Endometrioid233.45%00.00%36.00%204.50% Clear cell223.30%00.00%48.00%184.05% Mucinous182.70%00.00%36.00%153.38% Unspecified7811.72%2413.26%48.00%5111.49%Family history Yes497.36%3016.57%48.00%163.60%0.204.66e-080.26 No61792.64%15183.43%4692.00%42896.40%Chi-squared test was used to compare clinical characteristics between groups. A p-value of < 0.05 was considered as statistically significant. BRCA1/2^+^ patients are the ones with BRCA1 or BRCA2 P/LPVs; DDR^+^ patients are the ones with germline P/LPVs in non-BRCA1/2 DDR genes; DDR^−^ patients are the ones without any P/LPVs in DDR genes
Regarding histological subtypes, high-grade serous carcinoma (HGSC) was the predominant subtype, accounting for 75.8% (505/666) of the cases. Other subtypes included endometrioid (3.5%, 23/666), clear cell (3.3%, 22/666), low-grade serous ovarian cancer (3.0%, 20/666), and mucinous carcinoma (2.7%, 18/666), with 11.7% (78/666) cases unspecified. BRCA1/2^+^ patients showed a significant enrichment of HGSC compared to the DDR^+^ (p = 3.26e-07) and DDR^−^ (p = 1.04e-05) groups, except for one BRCA2^+^ patient presenting with low-grade serous ovarian cancer. Additionally, a family history of cancer was reported in 49 of the 666 patients (7.4%), with BRCA1/2^+^ patients showing a significantly higher likelihood compared to DDR^-^ patients (p = 4.7e-08).
Genetic predispositions in DDR genes
Each sample generated approximately 3 Mb of high-quality reads, with an average coverage exceeding 500×. Over 99% of the targeted regions achieved a coverage depth greater than 100×. A total of 232 P/LPVs spanning 21 genes were identified in 222 of the 666 patients. Notably, 12.9% (30/232) of these variants were previously unreported in ClinVar or other public databases (Table S2). The identified P/LPVs consisted of 142 frameshift, 57 stop-gain, 12 missense, 19 splice-site, and 2 start-codon variants.
Among the 19 splice-site variants, 14 were located at canonical acceptor/donor sites, 3 at intronic boundaries, and 2 at exon ends. One notable variant, BRCA1:c.132 C > T (p.Cys44=), a synonymous variant at the exon3 boundary, was detected in two unrelated individuals. Functional analyses revealed that this synonymous change disrupts protein function by inducing aberrant splicing [20].
The top five mutants identified in HOC patients were BRCA1 (n = 139), BRCA2 (n = 43), RAD51D (n = 11), MUTYH (n = 6), and RAD50 (n = 4), accounting for 59.9%, 18.5%, 4.7%, 2.5%, and 1.7% of all detected variants, respectively. Additional mutations were identified in GEN1 (n = 3), ATM (n = 3), BRIP1 (n = 3), FANCA (n = 3), RAD51C (n = 3), MRE11A (n = 2), MSH2 (n = 2) and MSH6 (n = 2) (Fig. 1). P/LPVs in FANCL, FANCM, PALB2, TP53, ATR, CHEK1, ERCC3, and RAD54L were each detected in only one patient. In general, most P/LPVs were mutually exclusive; however, ten patients harbored dual variants, consistently involving at least one in BRCA1 or BRCA2 (Table S3). This underscores the critical role of BRCA1/2 as key drivers in ovarian cancer pathogenesis.
Fig. 1. Genetic predispositions of pathogenic or likely pathogenic (P/LPVs) in Henan ovarian cancer (HOC) patients. Oncoplot presents the distribution of germline P/LPVs detected in ≥ 2 independent individuals of this study. The upper panel shows the number and subtype of PVs/LPVs detected in each patient. The middle panel displays the number of P/LPVs detected in each gene and its proportion among the total variants (n = 232). Clinical characteristics of the patients are indicated by different colors and clustered according to disease stages
Risk estimates of DDR genes in the HOC cohort
As expected, BRCA1 and BRCA2 demonstrated the strongest associations with ovarian cancer risk in our cohort. When compared with gnomAD, BRCA1 P/LPVs showed a significantly increased OR of 125.5 (95%CI: 88.7–180.1, p-adj = 2.0e-176), while BRCA2 P/LPVs had an OR of 17.9 (95%CI: 12.0–26.4, p-adj = 2.3e-33). A similar trend was also observed when compared with ChinaMAP database, where BRCA1 and BRCA2 P/LPVs showed ORs of 146.1 (95% CI: 89.4–253.1, p-adj = 7.6e-153) and 20.2 (95%CI: 12.5–32.6, p-adj = 1.2e-31), respectively.
Although less frequent, P/LPVs in RAD51D, RAD51C, and MSH2 also showed significantly increased ORs (OR > 10 and p < 0.05 for all) when compared with both the gnomAD and ChinaMAP databases. MUTYH, however, only exhibited significantly increased ORs when compared with the gnomAD database, as detailed in Table 2. For other DDR genes—including BRIP1, ERCC3, RAD54L, FANCL, TP53, MRE11A, FANCM, MSH6, RAD50, ATM, and FANCA—the ORs exceeded 2, but their associations with OC risk did not reach statistical significance (Table 2 and Table S4). This is likely due to their low variant prevalence and the limited sample size, which precluded precise risk estimation for these rare mutations in this study.
Table 2. Genes associated with increased ovarian cancer risk identified in Henan Ovarian Cancer (HOC) cohortGene listHOC (n = 666)gnomAD v4.1.0 East Asian(n = 22,448)ChinaMAP v2020-03.beta (n = 10,588)cases%cases%OR95% CIp-valuep-adjcases%OR95% CIp-valuep-adj BRCA1 13920.87%470.21%125.588.7–180.12.2e-1772.0e-176190.18%146.189.4–253.18.4e-1547.6e-153 BRCA2 436.46%860.38%17.912.0–26.45.1e-342.3e-33360.34%20.212.5–32.62.6e-321.2e-31 RAD51D 111.65%330.15%11.45.2–23.33.4e-081.0e-0750.05%35.411.3–130.29.9e-113.0e-10 RAD51C 30.45%100.04%10.11.8–39.65.5e-030.0130.03%15.92.1–119.43.6e-038.2e-03 MSH2 20.30%60.03%11.31.1–63.10.020.0320.02%15.91.2–219.10.020.04 MUTYH 60.90%620.28%3.31.2–7.60.010.02440.42%2.20.8–5.10.10.1 BRIP1 30.45%270.12%3.80.7–12.30.050.1160.15%3.00.6–10.50.10.2 RAD50 40.60%480.21%2.80.7–7.70.060.07320.30%2.00.5–5.60.20.2 MRE11A 20.30%170.08%4.00.4–16.80.10.150.05%6.40.6–38.90.060.09Fisher’s exact test was applied to determine relative risk for each gene by comparing to its prevalence in the HOC cohort to that in gnomAD (East Asian, v4.1.0) and ChinaMAP databases (v2020-03.beta). False discovery rate (FDR)-adjusted p-values were calculated using the Benjamini-Hochberg procedure. A p-value of < 0.05 was considered as statistically significant. OR: odds ratio; 95% CI: 95% confidence interval; p-adj: adjusted p-value
Discussion
In this study, we analyzed germline variants in 45 DDR genes among 666 Han Chinese OC patients from Henan, China, identifying P/LPVs in 33.3% (222/666) of the cases. BRCA1, BRCA2, RAD51D, RAD51C and MSH2 were confirmed as high-risk OC predisposition genes in Han Chinese (OR > 10, p < 0.05 for all). The prevalence of DDR gene P/LPVs in our cohort is higher than that reported in North American and European patients (20.5%–27.5%, Table S5) [21–23]. While the overall distribution of P/LPVs identified in this study largely mirrors patterns observed in other populations, there are some notable differences. For instance, mismatch repair gene variants were limited to MSH2 and MSH6 (0.6% vs. 1–2% in other studies) [21–26], likely due to the small number of patients with endometrioid carcinoma (n = 23).
Notably, BRCA1 contributed predominantly to the elevated prevalence of DDR gene predispositions observed in our HOC cohort. While established factors such as ethnicity, histologic subtype, and family history are known to influence BRCA1 prevalence, they do not fully account for the higher rate observed in our HGSC patients (31%, 156/505), which exceeds previous reports of up to 25% [22, 27, 28]. A review of domestic studies revealed substantial heterogeneity in BRCA1 prevalence, even among studies conducted by the same research group (Table S6) [11, 29–32], suggesting the presence of population-specific influences. Further analysis revealed a higher frequency of recurrent variants (RVs)—defined as the same P/LPV identified in three or more patients—indicating a potential founder effect (Table S7). This was supported by short tandem repeat (STR) analysis [11, 33], which showed that patients harboring RVs frequently shared identical STR haplotypes flanking the BRCA1 locus (Fig. S1). For instance, individuals with BRCA1:c.5470_5477del shared haplotypes D17S1320 (172) and D17S1327 (129), while those with BRCA1:c.5521delA shared D17S846 (237) and D17S1789 (193).
Although no significant difference in disease onset age was observed among BRCA1/2^+^, DDR^+^, and DDR^−^ patients (Table 1), cumulative incidence curves revealed that BRCA1^+^ patients experienced significantly earlier disease onset than the others (p < 0.01 for all; Fig. S2A), consistent with previous reports [34, 35]. Interestingly, DDR^−^ patients exhibited a higher incidence rate than BRCA1/2^+^ and DDR^+^ patients before the age of 40, suggesting that additional factors may contribute to early-onset disease in this subgroup.
The Androgen Receptor (AR) gene, expressed in all OC subtypes, harbors a CAG-repeat in exon1 that has been associated with disease onset age in breast and ovarian cancer [36, 37]. In HOC cohort, we identified 28 distinct AR-CAG alleles varying in size between 15 and 29 repeats, with 22-repeat being dominant that 88% (585/666) were AR-CAG^22/22^ homozygotes (Fig. S2B). Disease incidence curves showed that non-CAG^22/22^ patients were more likely to develop OC before age 40 (p = 0.0129, Fig. S2C). This trend was also observed in BRCA1/2^−^ patients (Fig. S2D and E), suggesting a *BRCA1/2-*independent effect. Prior studies on CAG-repeat length yielded inconsistent results [37–39]; it remains to be clarified whether disease risk is linked to repeat length or genotype. Of note, no similar trend was observed for the GCG-repeat at the same AR exon (Fig. S2F and G).
Our findings on high-risk OC genes (BRCA1/2, RAD51C/D, and MSH2; OR > 10 and p < 0.05) generally align with Western studies but differ for moderate- to low-risk genes. For instance, although MUTYH showed significance in comparison with gnomAD, its clinical relevance remains uncertain, as half of the six carriers also harboured BRCA1/2 PVs. Similarly, although BRIP1 showed borderline significance when compared with the gnomAD database, the association was no longer statistically significant after Benjamini–Hochberg correction for multiple testing. The lack of statistical significance for other moderate- to low-risk genes is likely attributable to the limited sample size of our HOC cohort, which may have reduced the power to detect modest effect sizes. Larger studies or pooled analyses will be necessary to clarify the contributions of these genes to ovarian cancer risk in our population. These results underscore the need for caution when interpreting moderate- to low-risk genes, as genetic susceptibility may be highly context-dependent [21, 22, 40–43].
The NCCN v2.2025 guideline list ten OC risk genes, including BRCA1/2, RAD51C/D, BRIP1, PALB2, MLH1, MSH2, EPCAM, and MSH6. However, UK experts recently noted that OC risk associated with RAD51C/D, BRIP1, and PALB2 strongly depends on family history, highlighting the need for personalized genetic counseling [44, 45]. In contrast, current Chinese guidelines still lack population-based risk evidence, which limits the accuracy and applicability of genetic risk assessment [46, 47]. There is an urgent need for Chinese-specific frameworks to support evidence-based counseling and preventive strategies.
This is the first study providing precise risk estimates for DDR genes in a relatively homogeneous Han Chinese cohort from central China, reducing geographic and ethnic confounding. This homogeneity enabled the identification of a strong BRCA1 founder effect, which partially explains the high BRCA1 prevalence in our cohort. However, limitations of our study include single-center design and small sample size, which restrict conclusions for moderate- to low-risk genes, e.g., MUTYH and BRIP1. Also, our panel (45 genes) did not detect large genomic rearrangements, possibly underestimating variant prevalence [48, 49]. Although formal gene burden testing was not performed, we examined genotype–phenotype associations within the HOC cohort by comparing clinical features across BRCA1/2^+^, DDR^+^, and DDR^−^ groups. This group-based comparison—conducted using chi-squared tests as described in the Methods section—provided a preliminary overview of potential genotype-related clinical differences. However, individual gene-level burden testing was not feasible due to the low number of carriers in most non-BRCA genes. The analysis requires larger cohorts to achieve adequate statistical power. Future studies with expanded patient numbers or collaborative meta-analyses will be essential to validate gene–phenotype associations more comprehensively.
Recognition of the BRCA1 founder effect in the Han Chinese population provides an opportunity to streamline genetic screening through targeted testing of recurrent variants. This approach can significantly reduce costs and improve feasibility for large-scale screening programs, particularly in resource-limited regions. In this context, our identification of recurrent variants such as BRCA1:c.5470_5477del supports the design of population-specific, first-tier panels tailored to Han Chinese patients. Moreover, our gene-specific risk estimates reveal striking ancestry-related differences. For example, RAD51D showed an OR of 35.4 in the Han Chinese cohort (vs. 6–12 reported in Western populations [44]), highlighting the importance of ethnicity-specific risk stratification. These findings argue against the direct application of Western-derived risk estimates to Chinese populations and underscore the need to develop China-specific clinical guidelines for genetic counseling and risk-reducing interventions. However, we also caution that our findings may not generalize to non-Han ethnic groups within China (e.g., Tibetan, Uyghur, Mongolian), for whom genomic data remain scarce. Without ancestry-matched reference data and founder variant characterization in these populations, extrapolation from Han Chinese data is inappropriate. We strongly advocate for inclusive, multi-ethnic research efforts to fill these knowledge gaps and support equitable clinical decision-making across China’s diverse population.
Conclusion
BRCA1/2, RAD51C/D, and MSH2 are high-risk OC predisposition genes identified in Han Chinese.
Supplementary Information
Supplementary Material 1.
Supplementary Material 2.