Occurrence of diverse circoviruses in wild birds in Hungary
Anna Pataki, Dóra Máté, Eszter Kaszab, Krisztina Bali, Krisztina Ursu, Enikő Fehér

TL;DR
This study found diverse circoviruses in wild birds in Hungary, suggesting they may spread widely and affect bird health and conservation.
Contribution
The study identifies new circovirus species and expands the host range of known circoviruses in wild birds.
Findings
Circoviruses were detected in 32 out of 588 wild bird samples.
New circovirus species were identified, including long-eared owl-associated CV1 and barn owl-associated CV1.
Known circoviruses were found in bird species previously not associated with them, such as white storks and peregrine falcons.
Abstract
Circoviruses (CVs) infect a wide range of avian hosts, including domestic, ornamental and wild birds. The immunosuppressive effect of these viruses could make the host more susceptible to other pathogens. This study aimed to assess the potential hosts and genetic features of CVs or other related viruses in wild birds. Cloacal swab samples of 588 birds were processed for screening of circoviral sequences using nested PCR. Altogether, 19 complete genome and 21 partial sequences of small, circular, rep-encoding DNA viruses were determined, 32 of which belonged to CVs. Along with some newly established CV species (virus name long-eared owl-associated CV1 and barn owl-associated CV1), genomic sequences of previously characterized avian CVs (pigeon CV, duck CV, goose CV, gull CV, swan CV and little bittern CV) were identified. Pathogenic CVs and CVs of unknown aetiology occur in wild birds…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
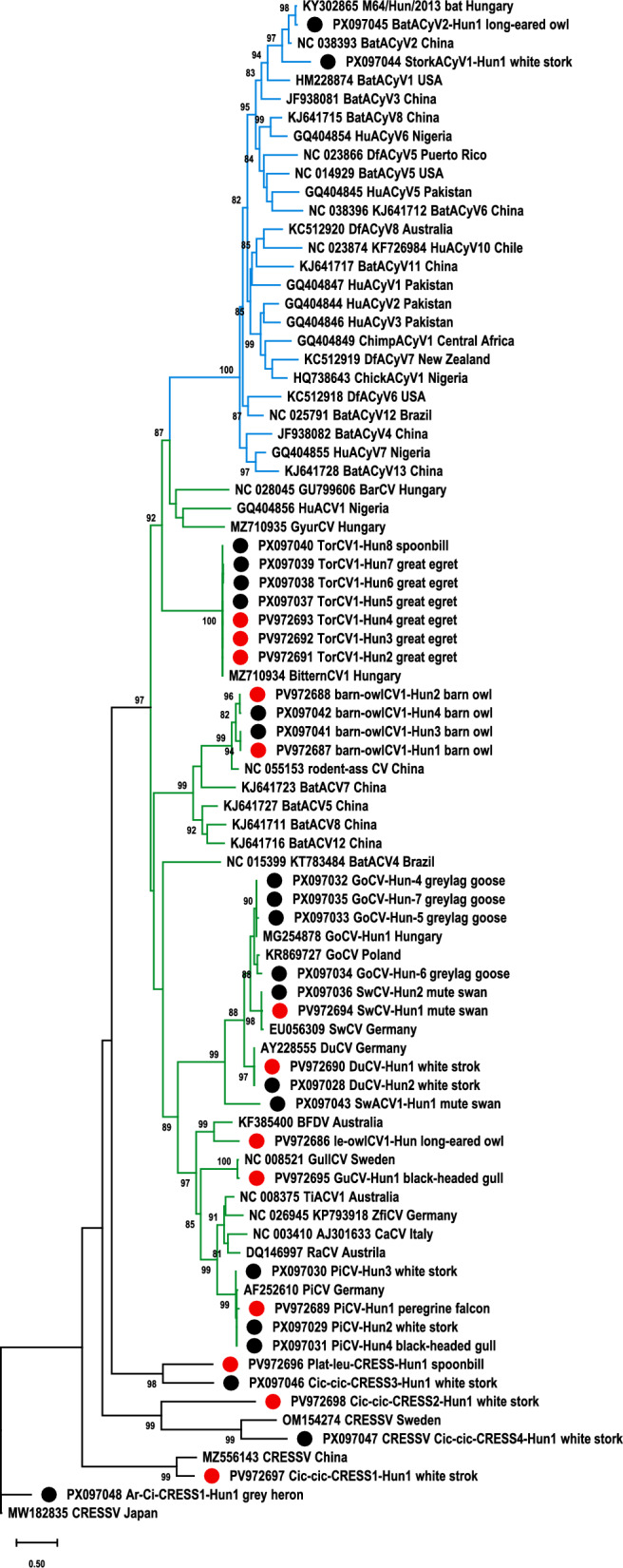 Figure 1
Figure 1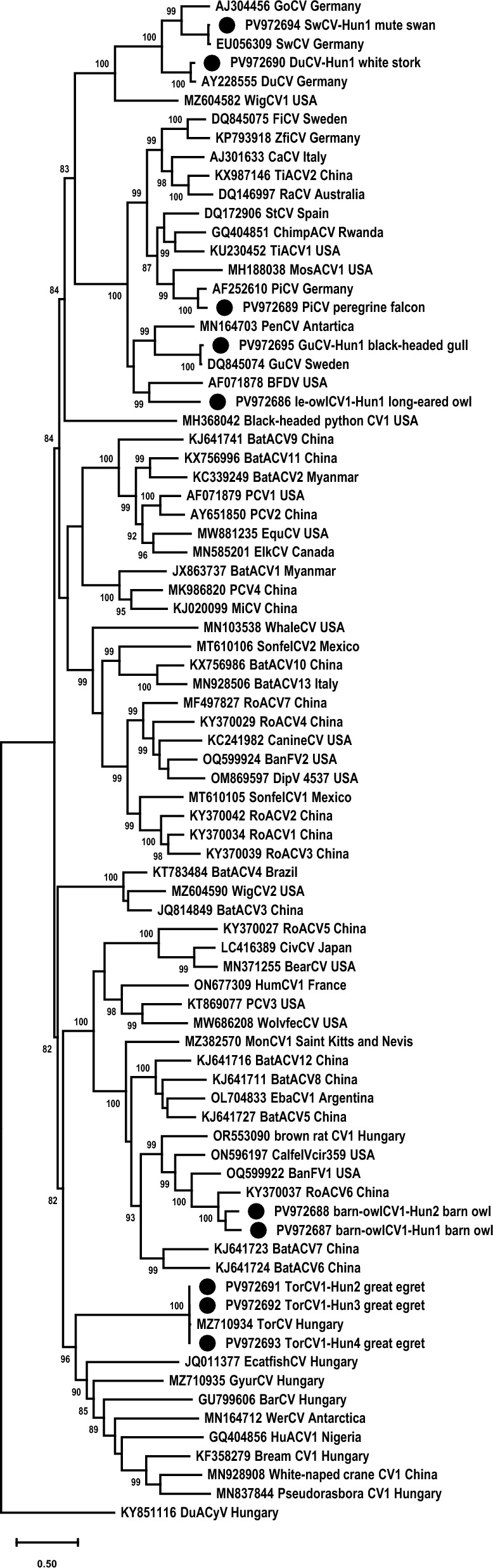 Figure 2
Figure 2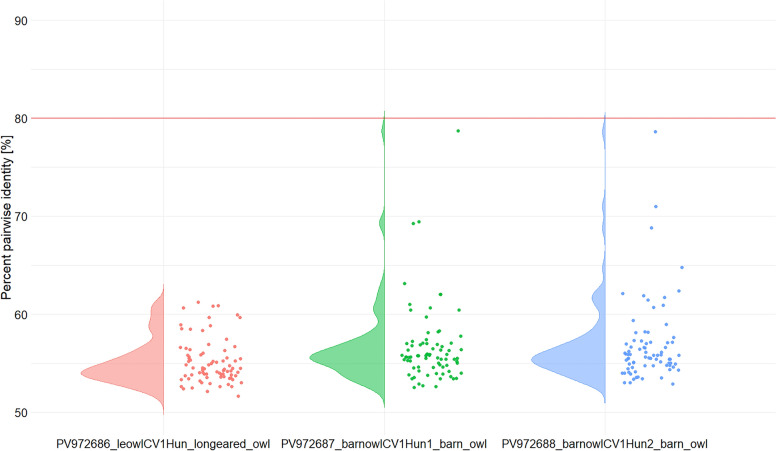 Figure 3
Figure 3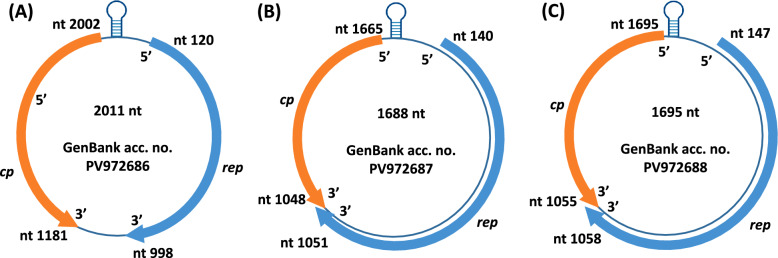 Figure 4
Figure 4- —Ministry of Culture and Innovation of Hungary from the National Research, Development and Innovation Fund, financed under the 2024-2.1.1-EKÖP funding scheme
- —National Research, Development and Innovation Office
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAnimal Virus Infections Studies · Virology and Viral Diseases · Respiratory viral infections research
Introduction
In relation to poultry production, a fundamental concern is that wild birds pose a threat to farmed animals. Indeed, wild birds can transmit some pathogens, such as influenza and Newcastle disease virus, through droppings, respiratory droplets and plumage, contaminating the feed, water and environment [1–4]. If spillover occurs from wild birds into domestic flocks, in cases where domestic birds are kept in high density, pathogens can rapidly spread. Disease outbreaks can lead to economic losses through slow growth rates, reduced egg production and increased mortality in poultry flocks [1, 3] However, attention must be also paid to the processes by which potential pathogens are released into the wider environment from domestic to wild animals [1, 2]. Even if a pathogen does not actively replicate within a wild bird, the animal may serve as a transmitter. The pathogen may spillover to novel hosts through shared resources and spread quickly by long-distance avian movements [1–3]. Agricultural intensification, the sacrifice of natural habitats for meat and feed production, makes wild bird populations increasingly vulnerable [1, 2].
Circoviruses (CVs; Circovirus genus, Circoviridae family, Cressdnaviricota phylum) possess small, circular, single-stranded DNA genomes of ~1600–2200 bases in length, with an ambisense arrangement in respect of the open reading frames of the replication-associated (Rep) and capsid (Cp) proteins [5, 6]. Cycloviruses (CyVs), members of the Cyclovirus genus of the Circoviridae family, have similar genomic properties, but the rep is located on the complementary strand compared to that of CVs [5–7]. CVs and CyVs have been detected in a diverse range of hosts, including mammals, birds, fish and insects, as well as in environmental samples such as water and soil [5–22]. Several other related small, circular, Rep-encoding single-stranded (CRESS) DNA viruses have been identified; however, data remain very limited regarding their host range, geographic distribution, genetic variability and tissue tropism. This is complicated by the fact that vast majority of these viruses cannot be propagated [23, 24].
Of the viral groups mentioned here, the pathogenic role of some CVs has been confirmed. Porcine CV2 and porcine CV3, beak and feather disease virus (BFDV), pigeon CV (PiCV), goose CV (GoCV), duck CV (DuCV), finch CV and canary CV have been associated with severe diseases in their hosts [25–35]. In general, CV infection of birds may lead to lethargy, weight loss, feathering disorders, beak deformities, diarrhoea and internal organ failure [27–31, 33, 34]. CV can induce immunosuppression; thus, the host may become susceptible for secondary infections and opportunistic pathogens, which can cause more severe forms of illnesses [27–31, 33, 34].
The aim of the study was to survey CVs and their potential host range among wild birds, as well as genetic characterization of the identified viruses. In total, complete genomes of 16 CVs and 3 other CRESS DNA viruses were determined, together with 21 partial sequences. A diverse picture in terms of host species was also obtained, confirming that some CVs may be widespread in the wild.
Materials and methods
Sample preparation and PCR
In total, 588 cloacal swab samples, collected for influenza virus surveillance throughout Hungary, were provided by the Veterinary Diagnostic Directorate, National Food Chain Safety Office, Budapest, Hungary. Sampled wild birds and sites of collection are listed in Additional file 1, while Additional file 2 includes the sites of collection of avian samples in which CRESS DNA virus sequences were identified. The nucleic acid was extracted with the KingFisher Flex system (ThermoFisher, Waltham, Ma, USA) using the MagAttract 96 cador Pathogen Kit (Qiagen, Hilden, Germany) according to the manual.
Broad-spectrum nested PCR was used for CV detection described by Li et al. [7] with the primers CV-F1 (5′-GGIAYICCICAYYTICARGG) and CV-R1 (5′-AWCCAICCRTARAARTCRTC) in the first PCR, and with CV-F2 (5′-GGIAYICCICAYYTICARGGITT) and CV-R2 (5′-TGYTGYTCRTAICCRTCCCACCA) for the second PCR [7]. The PCR mixtures contained 1× DreamTaq Green buffer, 250 μM of dNTP mix, 250 nM of primers, 0.5 U of DreamTaq DNA Polymerase (Thermo Fisher Scientific, Waltham, MA, USA) and 1 μl of nucleic acid templates in 20 μl final volume. The cycling protocol of the PCR was as follows: denaturation at 95 °C for 3 min; 40 cycles of denaturation at 95 °C for 30 s, 52 °C or 56 °C annealing (for the first and second PCR, respectively) for 30 s, and elongation at 72 °C for 1 min; final extension at 72 °C for 10 min. PCR products of ~400 bp in length were purified from agarose gel with Geneaid Gel/PCR DNA Fragments Extraction Kit (Geneaid Biotech, Taipei, Taiwan) and were directly sequenced with the CV-F2 and CV-R2 primers.
Inverse PCR primers were designed based on the results of the direct sequencing and were used in variable combinations for the amplification of the undetermined genomic regions of the respective virus (Additional file 3). If multiple products were generated for a sample, they were submitted for next-generation sequencing as a mixture. The PCR mixture contained 1× Phusion Green HF buffer, 200 μM of dNTP mix, 200 nM of primers, 3% DMSO, 0.25 U of Phusion DNA Polymerase (Thermo Fisher Scientific, Waltham, MA, USA) and 2 μl of the extracted nucleic acid in 25 μL final volume. The cycling protocol of PCRs was as follows: denaturation at 98 °C for 30 s; 45 cycles of denaturation at 98 °C for 10 s, annealing for 30 s at a temperature calculated for each primer, and elongation at 72 °C for 2 min; final extension at 72 °C for 5 min. The PCR products were purified from agarose gel with Geneaid Gel/PCR DNA Fragments Extraction Kit (Geneaid Biotech, Taipei, Taiwan) and were prepared for next-generation sequencing.
Next-generation sequencing
DNA libraries were prepared with the Illumina Nextera XT DNA Library Preparation Kit (Illumina, San Diego, CA, USA) combined with the Nextera XT Index Kit v2 Set C (Illumina, San Diego, CA, USA) [18]. Libraries were pooled and loaded into iSeq 100 i1 reagent cartridge v2, and were sequenced using the Illumina iSeq100 platform (Illumina, San Diego, CA, USA).
Bioinformatic analysis
The quality of raw reads was checked with the FastQC tool [36]. Based on the results, ten bases were trimmed from both the 3′ and 5′ ends of the reads. De novo assembly of the sequence reads occurred with the Tadpole assembler of the Geneious Prime software v.2025.1.2 (Biomatters Ltd., Auckland, New Zealand), and the contigs were identified with the Basic Local Alignment Search Tool (BLAST). Mapping to the reference (Geneious assembler and default parameters) was performed using the closest references found based on BLAST analysis (Additional file 4). Raw reads were remapped to the assembled genomes for another checking and the final contigs were merged with the sequences obtained by nested PCR and Sanger sequencing. Open reading frames were predicted with the Open Reading Frame Finder online software and were checked through BLAST analysis. Sequences were edited with Aliview and were aligned with the MAFFT algorithm implemented in Geneious Prime software [37]. Phylogenetic analyses were carried out with the PhyML 3.0 online tool, the best calculated models and SH-like branch support [38]. The phylogenetic trees were visualized and edited with the MEGA X software [39]. Pairwise nucleotide (nt) and amino acid (aa) identities were calculated and compared with Geneious Prime and SDTv1.2 software according to Varsani et al. using CV reference sequences found by BLAST search (Table 1) [40, 41]. The pairwise identities generated by the SDTv1.2 were imported into the RStudio environment (R version 4.5.1; RStudio version 2025.5.1.513) [42, 43]. The visualization of the data in a raincloud plot was performed using the packages ggplot2 package (v3.5.2), gghalves package (v0.1.4) and tidyverse package (v2.0.0) [44–46]. Table 1CRESS DNA viruses detected in the sampled wild birdsVirus nameVirus strainGenBank accession noSampled birdRep aa identity with referencesHighest nt identity with referenceDuck circovirusDuCV-Hun1PV972690White stork100% with EF45115799.7% with PV649664*Duck circovirusDuCV-Hun2PX097028White stork100% with PV64966499.8% with PV649664*Pigeon circovirusPiCV-Hun1, n = 2*PV972689Peregrine falcon98.1% with PV45439397.7% with KF738870*Pigeon circovirusPiCV-Hun2PX097029White stork99.2% with KX10878699.2% with OR801818Pigeon circovirusPiCV-Hun3PX097030White stork99.2% with OR99926598.9% with OR999239Pigeon circovirusPiCV-Hun4PX097031Black-headed gull100% with MW18195297.8% with DQ915950Goose circovirusGoCV-Hun4PX097032Greylag goose100% with OK07080399.8% with OK070803Goose circovirusGoCV-Hun5PX097033Greylag goose98.5% with OK07080399.3% with OK070803Goose circovirusGoCV-Hun6PX097034Greylag goose95.0% with KT80865792.9% with KR869727Goose circovirusGoCV-Hun7PX097035Greylag goose100% with OK07080399.8% with OK070803*Swan circovirusSwCV-Hun1, n* = 6*PV972694Mute swan98.6% with EU05630998.5% with EU056309*Swan circovirusSwCV-Hun2PX097036Mute swan98.5% with EU05630997.1% with EU056309*Gull circovirusGuCV-Hun1**PV972695Black-headed gull98.3% with JQ68585498.9% with JQ685854Little bittern circovirusTorCV1-Hun2PV972691Great egret100% with MZ71093499.4% with MZ710934Little bittern circovirusTorCV1-Hun3PV972692Great egret100% with MZ71093499.6% with MZ710934Little bittern circovirusTorCV1-Hun4PV972693Great egret100% with MZ71093499.6% with MZ710934*Little bittern circovirusTorCV1-Hun5PX097037Great egret100% with MZ710934100% with MZ710934Little bittern circovirusTorCV1-Hun6PX097038Great egret99.24% with MZ71093499.7% with MZ710934Little bittern circovirusTorCV1-Hun7PX097039Great egret100% with MZ71093499.7% with MZ710934Little bittern circovirusTorCV1-Hun8PX097040Spoonbill100% with MZ710934100% with MZ710934*Long-eared owl-associated circovirus 1le-owlCV1-Hun1**PV972686Long-eared owl70.2% with KF38542157.7% with KF385421Barn owl-associated circovirus 1barn-owlCV1-Hun1PV972687Barn owl88.5% with NC_05515377.9% with NC_055153Barn owl-associated circovirus 1barn-owlCV1-Hun2PV972688Barn owl87.8% with NC_05515377.5% with NC_055153*Barn owl-associated circovirus 1barn-owlCV1-Hun3PX097041Barn owl92.1% with NC_05515385.4% with NC_055153Barn owl-associated circovirus 1barn-owlCV1-Hun4PX097042Barn owl89.9% with NC_05515385.1% with NC_055153Swan-associated circovirus 1SwACV1-Hun1PX097043Mute swan55.6% with KR86972764.7% with KR869727Stork-associated cyclovirus 1StorkACyV1-Hun1PX097044White stork73% with NC_03839374.2% with NC_038393Bat faeces-associated cyclovirus 2BatACyV2-Hun1PX097045Long-eared owl97.2% with KY30286594.6% with KY302865*Ciconia ciconia-associated CRESS DNA virus 1Cic-cic-CRESS1-Hun1**PV972697White stork66.6% with MZ55614358% with MZ556143Ciconia ciconia-associated CRESS DNA virus 2Cic-cic-CRESS2-Hun1PV972698White stork43.3% with MT20649146.3% with MT206491*Ciconia ciconia-associated CRESS DNA virus 3Cic-cic-CRESS3-Hun1PX097046White stork30% with MK94737140% with MK947371Ciconia ciconia-associated CRESS DNA virus 4Cic-cic-CRESS4-Hun1PX097047White stork56.5% with OM15427456.5% with OM154274*Platalea leucorodia-associated CRESS DNA virusPlat-leu-CRESS-Hun1**PV972696Spoonbill39.8% with OQ599924**71.7% with HQ638064*Ardea cinerea-associated CRESS DNA virus 1Ar-Ci-CRESS1-Hun1PX097048Grey heron75.4% with MW18283581% with MW182835The table represents the highest pairwise identity values to the closest GenBank reference sequences identified by BLAST search. The highest nucleotide identity values show genome-wide identities for complete genome sequences (italics), and identity of the replication-associated protein coding gene in case of partial sequences (normal lettering). Two pigeon circovirus, as well as six swan circovirus complete genome sequences were identical and were detected in samples of the same avian species for the respective virus, thus were assigned with one GenBank accession number (PV972689 for the pigeon circovirus and PV972694 for the swan circovirus). aa, amino acid; nt, nucleotide; Rep, replication-associated protein.
Results
Sanger sequencing confirmed amplification of CRESS DNA virus rep sequences in 40 of the 588 (6.8%) cloacal swab samples. According to BLAST and phylogenetic analysis of the partial Rep, 32 aa sequences clustered with CVs and two with CyVs, while six were distantly related to unclassified CRESS DNA viruses (Table 1, Figure 1).Figure 1Unrooted, maximum likelihood phylogenetic tree of CRESS DNA virus replication-associated protein (Rep) amino acid sequences, generated with the PhyML software, Q.pfam + R + F model, SH-like branch support. Branch support values <80 were hidden. The scale bar indicates substitutions per site. Circoviruses and cycloviruses are highlighted with green and blue branch colours, respectively. Partial Rep sequences are labelled with black circles, while red circles refer to viruses where complete genome sequences were determined.
Complete genome sequence of 19 viruses was determined combining sequences of the diagnostic and inverse PCR products. The references, used for read mapping, and statistical data (total and assembled read number, mean coverage) of the next-generation sequencing are listed in Additional file 4. Based on the structure of the complete genome, sequence comparisons and phylogeny, 16 of the viruses were found to belong to the Circovirus genus, while three were unclassified CRESS DNA viruses (Figure 2, Table 2) [41].Figure 2Maximum likelihood phylogenetic tree of complete genome sequence of circoviruses rooted to the reverse complement of the duck-associated cyclovirus genome sequence. The tree was generated with the PhyML software, GTR + R model, SH-like branch support. Branch support values <80 were hidden. The scale bar indicates substitutions per site. The novel strains were labelled with black circles.Table 2Location of the predicted open reading frames in the genome of circoviruses and unclassified CRESS DNA viruses identified in the present studyGenome lengthrep**cp5′ IR/IR upstream rep3′ IR/IR downstream repLong-eared owl-associated circovirus 1, PV9726862011 ntnt 120–998 (292 aa)nt 2002–1181 (274 aa)nt 2003–119 (228 nt)nt 999–1180 (182 nt)Barn owl-associated circovirus 1, PV9726871688 ntnt 140–1051 (303 aa)nt 1665–1048 (205 aa)nt 1666–139 (162 nt)-Barn owl-associated circovirus 1, PV9726881695 ntnt 147–1058 (303 aa)nt 1–1 and 1695–1055 (213 aa)nt 2–146 (145 nt)-Pigeon circovirus (n = 2), PV9726892042 ntnt 48–1001 (317 aa)nt 1996–1172 (274 aa)nt 1997–47 (93 nt)nt 1002–1171 (170 nt)Duck circovirus, PV9726901949 ntnt 55–933 (292 aa)nt 1893–1120 (257 aa)nt 1894–54 (110 nt)nt 934–1119 (186 nt)Little bittern circovirus, PV972691- PV9726931935 ntnt 52–999 (315 aa)nt 1858–1229 (209 aa)nt 1859–51 (128 nt)nt 1000–1228 (nt 229)Swan circovirus (n = 6), PV9726941783 ntnt 55–936 (293 aa)nt 1730–975 (251 aa)nt 1731–54 (107 nt)nt 937–974 (38 nt)Gull circovirus, PV9726952035 ntnt 107–1024 (305 aa)nt 1934–1197 (245 aa)nt 1935–106 (207 nt)nt 1025–1196 (172 nt)Ciconia ciconia-associated CRESS DNA virus 1, PV9726972313 ntnt 39–1112 (357 aa)nt 1193–1945 (250 aa)nt 1946–38 (406 nt)nt 1113–1192 (80 nt)Ciconia ciconia-associated CRESS DNA virus 2, PV9726981818 ntnt 181–1128 (315 aa)nt 1762–1262 (166 aa)nt 1763–180 (236 nt)nt 1129–1261 (133 nt)Platalea leucorodia-associated CRESS DNA virus, PV9726962298 ntnt 79–978 (299 aa)nt 2150–1059 (363 aa)nt 2151–78 (226 nt)nt 979–1058 (80 nt)The first nucleotide of the predicted nonanucleotide motif was considered as nt 1 of the genome. aa, amino acid; cp, capsid protein coding gene; nt, nucleotide; rep, replication-associated protein coding gene.
The PiCV (n = 2, from peregrine falcon, Falco peregrinus), DuCV (n = 1, from white stork, Ciconia ciconia), little bittern CV (TorCV; n = 3, from great egret, Ardea alba), swan CV (SwCV; n = 6, from mute swan, Cygnus olor) and gull CV (GuCV; n = 1, from black-headed gull, Chroicocephalus ridibundus) complete genome sequences represented high pairwise nt identities with reference sequences (Table 1, Figures 1 and 2). The PiCV and SwCV complete genome sequences obtained from this study were identical among each other, and originated from specimens of the same bird species, thus, these were referred with one accession number for the respective virus (PV972689 for the PiCV and PV972694 for the SwCV sequences). Three viruses formed two separate branches among CVs in the phylogenetic tree (Figure 2). The genomic sequence of the three strains (GenBank accession nos. PV972686, PV972687 and PV972688) showed ≤80% genome-wide pairwise identity with CV reference sequences of all species of the Circovirus genus (Table 1, Figure 3). In addition, the genomic structure of the here-characterized viruses is consistent with that of described for other CVs (Figure 4). Thus, these could be considered as members of new CV species [41]. One of the viruses, distantly related to BFDV, originated from long-eared owl (Asio otus) and was named long-eared owl-associated circovirus 1 (tentative species name Circovirus fulesbagoly, FulCV; GenBank accession no. PV972686) (Figure 2). The other two strains were grouped with the rodent-associated circovirus 6 and were listed as barn owl-associated circovirus 1 (tentative species name Circovirus gyongybagoly, GyoCV; GenBank accession nos. PV972687 and PV972688) after the sampled birds (barn owl, Tyto alba) (Figure 2). The two GyoCV strains showed 83.4% genome-wide identity with each other and had comparable genomic structure (Figure 4). The nonanucleotide motif upstream the rep, and Rep aa motifs of the novel CVs (RCR and Walker motif, motif C, Arg motif), as well as the arginine-rich N terminus of the Cp were characteristic of CVs (Table 3) [41]. The predicted cp of the FulCV, similarly to certain avian CVs (such as BFDV), started with alternative start codon [41].Figure 3Genome-wide pairwise identity values calculated for the novel circovirus sequences (long-eared owl-associated circovirus 1, strain le-owlCV1-Hun1, GenBank accession number PV972686 in red; barn owl-associated circovirus 1, strain barn-owlCV1-Hun1, GenBank accession number PV972687 in green; barn owl-associated circovirus 1, strain barn-owlCV1-Hun2, GenBank accession number PV972688 in blue) using reference sequences of all circoviruses (all circovirus species) and applying the matrix generated by SDTv1.2 software.Figure 4Schematic representation of the genomic structure of the long-eared owl-associated circovirus 1 (A), and the two strains of the barn owl-associated circovirus 1 (B and C).Table 3The nonanucleotide motif, as well as amino acid motifs of the replication-associated protein identified in the novel circovirus genomesLong-eared owl-associated circovirus 1, PV972686Barn owl-associated circovirus 1, PV972687 and PV972688Nonanucleotide motifTAGTATTACTAGTATTACRCR motif ICFTLNNCFTVNNRCR motif IIPHLQGPHLQGRCR motif IIIDNLKYCSKQNQTYCQKWalker-A motifGPPGCGKSRWASGPPGVGKTRYAVWalker-B motifVMDDYMLDDFMotif CITSNITSNArg finger motifALFRRFAMFRRI
Regarding the partial Rep aa sequences, 16 clustered with CVs and 2 with CyVs, while 3 with unclassified CRESS DNA virus sequences (Figure 1). Four of the partial CV sequences originated from greylag goose (Anser anser) and, based on the identity values, were amplified from GoCV genome, while others clustered with DuCV (n = 1, white stork) PiCV (n = 3, two from white storks and one from black-headed gull), TorCV1 (n = 4, three from great egret and one from spoonbill, Platalea leucorodia), SwCV (n = 1, mute swan) and GyoCV (n = 2, barn owl). The mentioned partial sequences represented high identity with corresponding references of the GenBank and/or the predicted rep of the here-determined complete genome sequences. DuCV sequences, obtained in this study, showed the highest identities with references originating from farmed ducks in China, South Korea and Thailand, while GoCV and PiCV sequences were closely related to those from Poland, as found by BLAST analyses. TorCV and SwCV have been described in wild birds in Hungary and Germany, respectively [11, 18]. Accession number of selected references chosen based on BLAST analysis are listed in Table 1.
One partial sequence, gained from mute swan, was distantly related (≤55.6% aa identity) with CVs, and was tentatively named swan-associated circovirus 1 (SwACV1). One of the two putative CyV sequences, originating from long-eared owl, showed high identity (94.6% nt, 97.2% aa) with bat faeces-associated CyV2 detected in Hungary (GenBank accession no. KY302865, Table 1). The other sequence, tentatively named stork-associated CyV1, was amplified from white stork and represented ≤73% aa identity with its closest related sequence, bat faeces-associated CyV2 from China (GenBank accession no. NC_038393, Table 1). Although both partial CyV sequences clustered with bat faeces-associated CyV2, these were only distantly related to each other (66.67% pairwise aa identity). Of note, the two bat faeces-associated CyV2 reference strains shared only 81.9% genome-wide identities. Partial Rep aa sequence of the other three, presumably unclassified CRESS DNA viruses (Ciconia ciconia-associated CRESS DNA virus 3 and Ciconia ciconia-associated CRESS DNA virus 4 from white storks and Ardea cinerea-associated CRESS DNA virus 1 from grey heron, Ardea cinerea) were distantly related to any reference sequences and were named also after the sampled bird (Table 1).
Discussion
Agricultural intensification, high density of livestock and the decline of natural habitats has led to increased loads of relevant pathogens in the environment, water and soil [1, 2]. This can increase the likelihood of synanthropic birds shedding microbes, whether they become infected or serve as simple transmitters. The consequences of microbes entering the wild is unknown, but it certainly poses a threat that, owing to the overlap of habitats, may affect to farming or pet aviaries [1–3]. On the other hand, as is known, some pathogens endangering poultry and ornamental birds originate from wild animals, including highly pathogenic avian influenza virus [47].
The augmented agricultural activity favours synanthropic species, such as house sparrow (Passer domesticus), columbids, crows, starling (Sturnus vulgaris) or magpie (Pica pica), but more birds are forced to adopt to the built environment [48, 49]. In contrast to the ‘typical’ synanthropic animals, the sampled wild birds are less likely to come into close contact with industrial and backyard farming, but their behaviour depends on the populated geographical region and the availability of resources [50]. The sampled species white stork, black-headed gull or barn owl could also visit or live near settlements [50]. DuCV and PiCV was identified not only in these wild birds, but PiCV was also amplified from peregrine falcon. DuCV and GoCV have been previously found in wild birds of the respective avian groups (Anseriformes), including a project in which GoCV was detected in birds of variable lifestyle in Hungary [20, 51–54]. The white stork is taxonomically distant from the host species described carrying DuCV in the literature [54, 55]. PiCV were identified in columbids, except a raven suffering from lymphoid depletion and opportunistic infection, as well as in zoo birds, where close cohabitation and the constant presence of rock dove facilitate spillover [56–58].
In a previous study, the TorCV sequence was identified in little bittern sampled in Hungary, while TorCV sequences in the present project were amplified from great egret and spoonbill cloacal samples [18]. As in the case of any viruses characterized here, the infection of birds could not be proven and it is possible that the viruses only passed through the gastrointestinal tract of the birds without infection and replication. Nevertheless, considering that TorCV has only been found in freshwater birds so far, it is conceivable that this group of birds are natural hosts. SwCV has also been diagnosed in aquatic birds, in organ samples of swan [11]. A high incidence of SwCV was indicated at the site in Germany, but no further data were available and these are the first sequences since [11].
Genomic sequences of the two novel CVs, FulCV and GyoCV, originated from Strigiformes, and represent the closest relation with rodent-associated viruses. Based on the demarcation criteria of the International Committee on Taxonomy of Viruses, these viruses can be considered to belong to a new species, as both are relatively distant from the closest references from a genetic point of view [41]. However, dietary origin introduced by the prey cannot be ruled out, as in case of bat faeces-associated CyV2 found in a long-eared owl. Of note, the bat faeces-associated CyV2 sequence was also amplified from faeces of the associated mammal. Unfortunately, only partial sequences could be determined for two additional, potentially new members of the Circoviridae family that were found in mute swan and white stork specimens (swan-associated circovirus 1 and stork-associated CyV1), but these could be identified after complete genome sequence determination.
Although countless genome sequences are available, the knowledge of unclassified CRESS DNA viruses is incomplete [23, 24]. Thus, it is very difficult to assess the results. Six distantly related viruses were classified as CRESS DNA viruses from white stork, spoonbill and grey heron samples. This group of viruses has often been identified in environmental samples, including water and soil [6, 24]. Thus, water may also play an intermediary role in this case as well.
A relatively large proportion of the samples examined were of white stork origin (Additional file 1). This shows that white stork may be a transmitter of CVs, CyVs and unclassified CRESS DNA viruses. The breeding range of these long-distance migratory birds extends from Europe to northwestern Africa, and western and central Asia. The Hungarian Plain, along with Mediterranean areas with rivers and lakes are ideal stopover and breading habitats [59, 60]. Waterfowls prefer these watery areas rich in resources as well, but countless other migratory and resident birds reside in wetlands, which facilitate the transmission and shedding of pathogens, including CVs [61]. GoCV and PiCV sequences, obtained in the present study from greylag goose and white stork for example, represented the highest identities with sequences originating from Poland, suggesting that these viruses may have been transferred to variable geographical regions by migratory birds [61].
In conclusion, addressing the issues of the wildlife–agriculture interface is of great importance to maintain their balance and to approach the essence of the One Health concept. In parallel to the necessary expansion of agriculture, we must also consider the preservation of wildlife. Of course, it is not sufficient to focus on a single highly pathogenic microbe, but we must take numerous factors into account to protect poultry and wild bird populations. Birds living in the vicinity of poultry farms can serve as a link between the built environment and wilderness. Although CVs have mostly indirect, less dramatic effect on the host organism than highly pathogenic microbes, their presence cannot be ignored. Regular monitoring of both poultry and wild bird pathogens and hosts allows early recognition and effective intervention.
Supplementary Information
Additional file 1. **The list of sampled wild birds and sites of sample collection.**Additional file 2. **Sites of collection of avian samples in which circular, Rep-encoding single-stranded DNA virus sequences were identified.**Additional file 3. **The list of the inverse primers used in this study. Letters F and R in the name of primers refer to the forward or reverse orientation of those.**Additional file 4. References used for read mapping and statistics of the next-generation sequencing.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Andrews S (2010). Fast QC: a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/. Accessed 9 Nov 2025
- 2R Core Team (2025) R: a language and environment for statistical computing. R foundation for statistical computing. https://www.R-project.org/. Accessed 31 Aug 2025
- 3Posit team (2025) Rstudio: integrated development environment for R. Posit software. https://www.posit.co/. Accessed 31 Aug 2025
- 4Magyar Madártani és Természetvédelmi Egyesület. Magyar madártani és természetvédelmi egyesület–hivatalos honlap. https://mme.hu/. Accessed 9 Nov 2025
- 5White Stork–an overview Science Direct topics. https://www.sciencedirect.com/topics/agricultural-and-biological-sciences/white-stork. Accessed 3 Sep 2025