Genetic Pigmentary Disorders: From Molecular Mechanisms to Clinical Manifestations
Ken Okamura, Tamio Suzuki

TL;DR
This review explores genetic disorders affecting pigmentation, explaining their molecular causes and clinical effects on skin, eyes, and other systems.
Contribution
The paper provides a comprehensive overview of both hypopigmentary and hyperpigmentary genetic disorders, linking molecular mechanisms to clinical outcomes.
Findings
Hypopigmentary disorders like oculocutaneous albinism result from mutations affecting melanin production and transport.
Hyperpigmentary conditions such as dyschromatosis symmetrica hereditaria involve distinct genetic pathways and clinical features.
RASopathies demonstrate how pigmentary abnormalities can co-occur with multisystem developmental issues.
Abstract
Genetic pigmentary disorders represent a diverse group of genetic conditions characterized by alterations in melanin production and transport and melanocyte development, resulting from single‐gene pathological variants. These disorders encompass both hypopigmentary and hyperpigmentary phenotypes, affecting not only skin pigmentation but also ocular, auditory, and systemic manifestations. This review examines the molecular mechanisms underlying major genetic pigmentary disorders, including hypopigmentary (e.g., oculocutaneous albinism, piebaldism, and Waardenburg syndrome) and hyperpigmentary (e.g., dyschromatosis symmetrica hereditaria, dyschromatosis universalis hereditaria, reticulate acropigmentation of Kitamura, and Dowling–Degos disease) disorders. Additionally, we discuss RASopathies, in which pigmentary abnormalities occur alongside multisystem developmental anomalies.…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
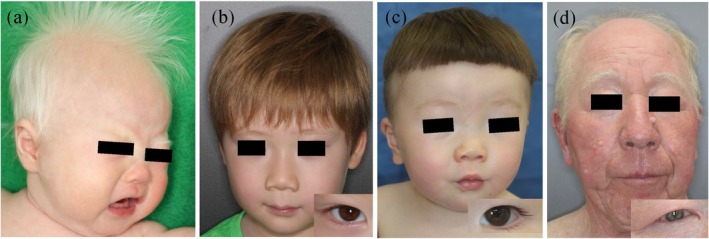 FIGURE 1
FIGURE 1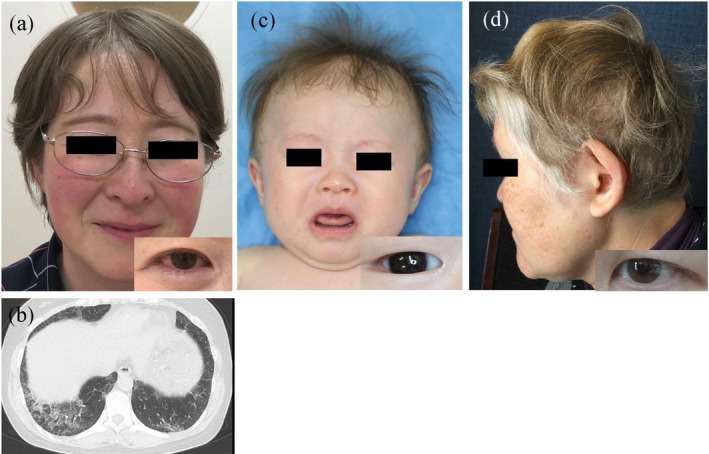 FIGURE 2
FIGURE 2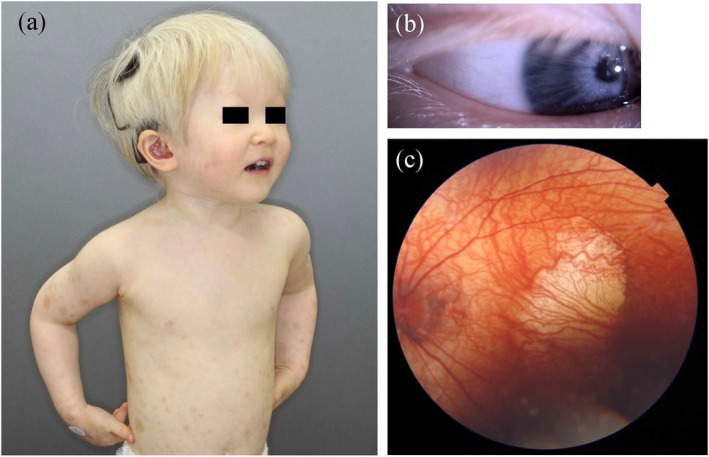 FIGURE 3
FIGURE 3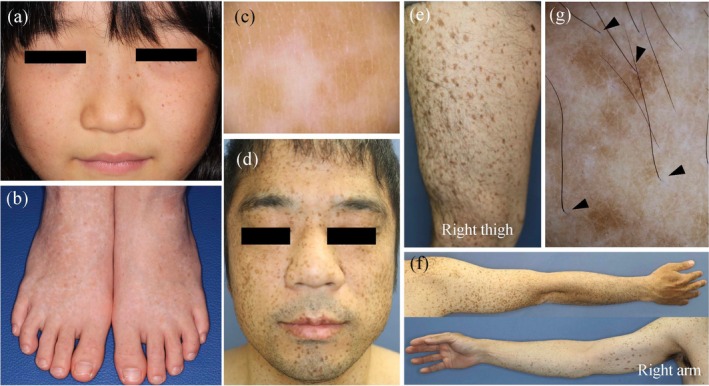 FIGURE 4
FIGURE 4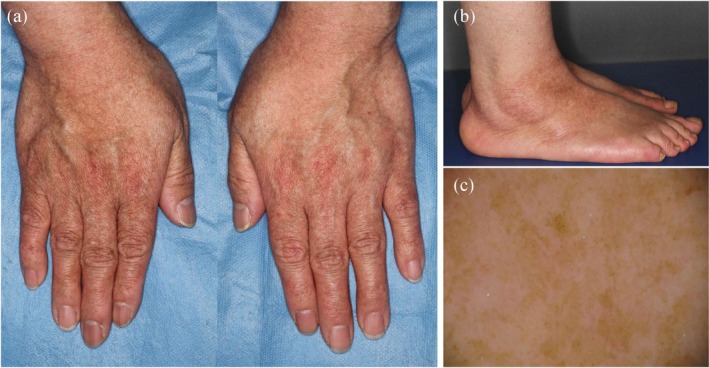 FIGURE 5
FIGURE 5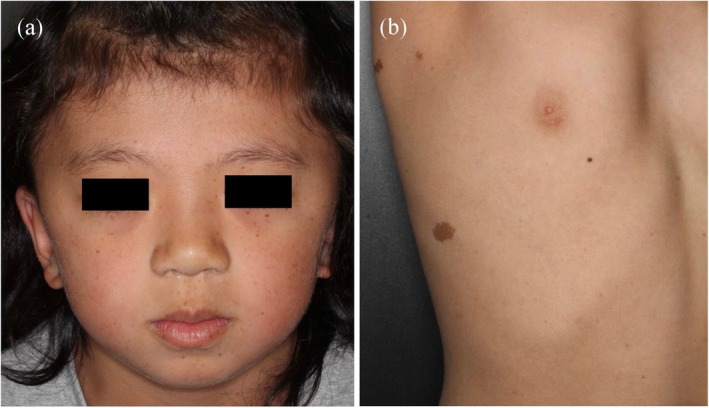 FIGURE 6
FIGURE 6| Phenotype (OMIM no.) | Gene (OMIM no.) | |
|---|---|---|
| 1. Nonsyndromic OCA | ||
| OCA1 |
| (*606933) |
| OCA1A (#203100) | ||
| OCA1B (#606952) | ||
| OCA2 (#203200) |
| (*611409) |
| OCA3 (#203290) |
| (*115501) |
| OCA4 (#606574) |
| (*606202) |
| OCA5 (%615 312) | 4q24 | |
| OCA6 (#113750) |
| (*609802) |
| OCA7 (#615179) |
| (*614537) |
| OCA8 (#619165) |
| (*191275) |
| 2. Syndromic OCA | ||
| a. Hermansky–Pudlak syndrome (HPS) | ||
| HPS1 (#203300) |
| (*604982) |
| HPS2 (#608233) |
| (*603401) |
| HPS3 (#614072) |
| (*606118) |
| HPS4 (#614073) |
| (*606682) |
| HPS5 (#614074) |
| (*607521) |
| HPS6 (#614075) |
| (*607522) |
| HPS7 (#614076) |
| (*607145) |
| HPS8 (#614077) |
| (*609762) |
| HPS9 (#614171) |
| (*604310) |
| HPS10 (#617050) |
| (*607246) |
| HPS11 (#619172) |
| (*607289) |
| b. Chediak–Higashi syndrome (#214500) | ||
|
| (*606897) | |
| 3. Others | ||
| Ocular albinism type 1 (#300500) | ||
|
| (*300808) | |
| FHONDA (#609218) |
| (*615585) |
| Clinical type | Clinical features | Genetic subtype/special type | Gene |
|---|---|---|---|
| WS1 | All three major features present |
| |
| WS2 | No facial abnormalities | WS2A |
|
| WS2E |
| ||
| WS2F |
| ||
| TADS |
| ||
| WS3 | Upper limb deformities |
| |
| WS4 | Associated with Hirschsprung disease | WS4A |
|
| WS4B |
| ||
| WS4C |
| ||
| PCWH |
|
- —Ministry of Health, Labor, and Welfare of Japan
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
Topicsmelanin and skin pigmentation · RNA regulation and disease · Connexins and lens biology
Introduction
1
Human skin pigmentation is a complex biological process primarily mediated by melanocytes, specialized neural crest‐derived cells residing in the skin epidermis and hair follicles, which synthesize and distribute melanin pigments within melanosomes, specialized lysosome‐related organelles (LROs) that function as the primary sites of melanogenesis [1, 2, 3]. Melanocyte development involves migration from the neural crest under the control of key signaling pathways, including the stem cell factor/c‐KIT, endothelin, Wnt, and α‐melanocyte‐stimulating hormone/melanocortin‐1 receptor pathways, with microphthalmia‐associated transcription factor serving as the master transcriptional regulator [4]. Melanin biosynthetic pathway begins with tyrosine conversion to DOPAquinone by tyrosinase (TYR), leading to eumelanin (brown–black) or pheomelanin (red–yellow) synthesis, followed by melanosome transport and melanin transfer to keratinocytes.
Pathological variants in genes regulating these processes result in genetic pigmentary disorders (GPDs) [5]. These disorders are broadly categorized into hypopigmentary disorders, including oculocutaneous albinism (OCA), piebaldism, and Waardenburg syndrome (WS), and various hyperpigmentary disorders, such as dyschromatosis symmetrica hereditaria (DSH), dyschromatosis universalis (DUH), reticulate acropigmentation of Kitamura (RAK), and Downling–Degos disease (DDD). Additionally, RASopathies represent an important group of disorders caused by germline variants in genes encoding components of the RAS–mitogen‐activated protein kinase (MAPK) signaling pathway, which manifest as diverse pigmentary phenotypes, including café‐au‐lait macules and lentigines, along with characteristic systemic developmental abnormalities. The clinical significance of GPDs extends beyond cosmetic concerns, as many conditions exhibit multisystem manifestations requiring comprehensive medical management [5].
Recent advances in genetic sequencing technologies have significantly enhanced our understanding of the genetic landscape of these disorders, revealing novel disease genes and elucidating complex genotype–phenotype relationships [6, 7, 8]. This review provides a comprehensive overview of the molecular basis, clinical features, and diagnostic approaches for major GPDs and emphasizes the importance of genetic testing for accurate diagnosis, patient management, and genetic counseling.
Hypopigmentary Disorders
2
OCA
2.1
OCA is an autosomal recessive genetic disorder characterized by the congenital reduction or absence of melanin in the eyes, skin, and hair due to pathogenic variants of the genes involved in melanin biosynthesis. It is broadly classified into nonsyndromic and syndromic subtypes. Nonsyndromic subtypes present with only pigment reduction and ocular symptoms, whereas syndromic subtypes are accompanied by other systemic complications [6]. Albinism can also present as ocular albinism, affecting only the eyes, or as foveal hypoplasia, optic nerve decussation defects, and anterior segment dysgenesis syndrome (FHONDA) [9]. Table 1 shows a list of OCA subtypes and their causative genes identified to date. Nonsyndromic OCA is caused by abnormalities in gene groups directly affecting melanin synthesis, with seven causative genes and one causative locus identified to date. In contrast, syndromic OCA subtypes, including Hermansky–Pudlak syndrome (HPS) and Chédiak–Higashi syndrome (CHS), are caused by abnormalities in genes involved in the biogenesis, trafficking, and function of LROs, including melanosomes. Eleven causative genes for HPS have been identified to date. The causative gene for CHS is LYST. In the following sections, we describe each nonsyndromic and syndromic OCA subtype in detail based on the statistical data of Japanese patients.
Nonsyndromic OCA
2.2
Nonsyndromic OCA includes eight reported subtypes: OCA1–8. In Japanese patients, OCA4 is the most common (23.1%), followed by OCA1 (19.7%) and OCA2 (10.3%) [10]. Additionally, only three cases of OCA3 and one case of OCA6 have been reported to date [11, 12, 13]. OCA1 is clinically further divided into OCA1A and OCA1B. Patients with OCA1A exhibit characteristic clinical features. Due to loss‐of‐function variants in the causative gene TYR, which encodes the rate‐limiting enzyme in melanin synthesis, they completely lack melanin‐synthesizing capacity, which results in white hair and milky white skin, with no pigment enhancement (tanning) upon UV exposure (Figure 1a). Ocular symptoms, such as nystagmus, photophobia, and amblyopia, are also severe, significantly reducing the patient's quality of life. In contrast, OCA1B retains some TYR activity; therefore, degree of pigment reduction is milder and ocular symptoms are often less severe in this subtype than in OCA1A (Figure 1b). Other subtypes also present varying degrees of severity depending on the variant type. Variant NM_000275.3:c.1441G>A (p.Ala481Thr) is present at an exceptionally high frequency in Japanese individuals with OCA2, whereas its allelic frequency is 0.12 in healthy individuals [14]. This variant retains approximately 70% of its function and is associated with the Japanese skin color [15]. Many Japanese patients with OCA2 are compound heterozygotes for the Ala481Thr variant and another pathogenic variant, typically showing mild clinical symptoms (Figure 1c). As mentioned above, OCA4 is the most common subtype among Japanese individuals, likely due to the high frequency of the loss‐of‐function variant NM_016180.5:c.469G>A (p.Asp157Asn) and presence of promoter variant NM_016180.5:c.‐492_489delAATG that retains some function in these individuals [16, 17]. The severity of OCA4 varies depending on the variant type and combination; however, patients with null variants, such as the homozygous Asp157Asn variant, exhibit severe pigment reduction and ocular symptoms (Figure 1d). The causative gene for OCA3 encodes TYRP1, a downstream enzyme in melanin synthesis specifically involved in eumelanin production, which generally results in mild clinical symptoms. In contrast, OCA6 is characterized by mild pigment reduction but severe ocular symptoms [13].
Clinical features of patients with various nonsyndromic oculocutaneous albinism (OCA) subtypes. (a) A 2‐month‐old girl with OCA1A presents with white to blond hair and light skin. She harbors compound heterozygous variants in TYR: NM_000372.5:C.929dupC (p.Arg311fs) and c.1292C>T (p.Pro431Leu). (b) A 6‐year‐old boy with OCA1B shows light brown hair and irises with compound heterozygous variants in TYR: NM_000372.5:C.230G>A (p.Arg77Gln) and c.1037‐7 T>A. He exhibits no nystagmus but shows mild amblyopia. (c) A 1‐year and 9‐month‐old boy with OCA2 presents with light brown hair and irises. He exhibits photophobia, but not nystagmus. He harbors compound heterozygous variants in OCA2: NM_000275.3:C.1182G>A (p.Met394Ile) and c.1441G>A (p.Ala481Thr). (d) A 74‐year‐old man with OCA4 born to consanguineous parents shows blond hair, gray irises, and severe ocular manifestations. He harbors a homozygous variant in SLC45A2: NM_016180.5:C.469G>A (p.Asp157Asn).
Syndromic OCA
2.3
HPS is a syndrome in which the membrane transport of proteins necessary for LRO function is impaired. These LROs include melanosomes in melanocytes, platelet‐dense granules in platelets, lamellar bodies in lung pneumocytes, lytic granules in cytotoxic T and natural killer cells, and other specialized organelles. HPS is caused by functional abnormalities in the protein complexes involved in membrane transport, with four protein complexes reported to date: Biogenesis of LRO complex (BLOC)‐1, BLOC‐2, BLOC‐3, and adaptor protein (AP)‐3. Clinical symptoms of HPS result from the impaired function of these protein complexes. Notably, HPS1 accounts for > 10% Japanese OCA cases [10], primarily explained by the founder variant NM_000195.5:c.398 + 5G > A in HPS1, which represents approximately half of all pathogenic variants in Japanese patients with HPS1 [6, 18]. HPS1, along with HPS4, constitutes the BLOC‐3 protein, which we refer to here as BLOC‐3 disease. Patients with BLOC‐3 disease present with severe pigment reduction and ocular symptoms and frequently develop fatal pulmonary fibrosis in adulthood (often after middle age; Figure 2a,b). Currently, lung transplantation is the only effective treatment [19]; however, novel therapeutic approaches, including gene therapy strategies, may be developed in the future. BLOC‐2 disease (HPS3, HPS5, and HPS6) is characterized by mild pigment reduction but severe ocular symptoms (Figure 2c) [6]. BLOC‐2 deficiency typically presents without severe systemic complications, with platelet aggregation function often remaining intact [20, 21, 22]. This preservation of function is possibly due to the position of BLOC‐2 downstream from BLOC‐1 in the cellular cargo transport pathway [23]. In contrast, patients consistently develop critical ocular abnormalities, including nystagmus and amblyopia. This clinical pattern suggests that BLOC‐2 plays a disproportionately important function in the retinal pigment epithelium compared to that in neural crest cell‐derived melanocytes. BLOC‐1 disease (HPS7, HPS8, HPS9, and HPS11) is extremely rare worldwide, with only one Japanese case of HPS9 reported to date (Figure 2d) [24]. The severity of OCA varies and is possibly associated with schizophrenia [24, 25]. AP‐3 disease (HPS2 and HPS10, caused by variants in AP3B1 and AP3D1, respectively) is characterized by immunodeficiency in addition to the classic HPS features. Both subtypes can develop interstitial pneumonia; however, HPS10 is distinguished by severe neurological manifestations, including seizures, neurodevelopmental delays, and hearing impairment. This neurological involvement in HPS10 is because AP3D1 encodes the δ subunit essential for both the ubiquitous and neuronal forms of the AP3 complex. In contrast, AP3β3A subunit affected in HPS2 is substituted by AP3β3B in the neuron‐specific heterotetramer, allowing neuronal AP3 function to be preserved in patients with HPS2 [26].
Clinical features of patients with various subtypes of the Hermansky–Pudlak syndrome (HPS). (a and b) A 44‐year‐old woman with HPS1 presents with light brown hair and hazel irises. She exhibits severe eye manifestations, including nystagmus and amblyopia. She harbors compound heterozygous variants in HPS1: NM_000195.5:C.1182delT (p.Leu395CysfsTer4) and c.2003 T>C (p.Leu668Pro). Computed tomography revealed pulmonary fibrosis. (c) A 9‐month‐old girl with HPS6 born to nonconsanguineous parents shows mild hypopigmentation and severe ocular manifestations, including nystagmus and amblyopia. She harbors a homozygous variant in HPS6: NM_024747.6:C.2038C>T (p.Gln680Ter). (d) A 52‐year‐old woman with HPS9 born to consanguineous parents presents with blond hair and light brown irises. She harbors a homozygous variant in BLOC1S6: NM_012388.4:C.285_286dupTC (p.His96LeufsTer22). She also presents with mild leukopenia, thrombocytopenia, and schizophrenia.
CHS develops due to the enlargement of LROs, characterized by giant granules in leukocytes and giant melanosomes in melanocytes, and dysfunction caused by functional abnormalities in the lysosomal trafficking regulator [27]. A distinctive clinical feature differentiating CHS from other OCA subtypes is the coexistence of hypopigmentation and hyperpigmentation following sun exposure, a phenomenon especially noticeable in Asian patients [28, 29]. Additionally, their hair characteristically exhibits a silver‐gray luster and is called “silver‐gray hair.” As it is accompanied by severe complications, such as immunodeficiency, central nervous system symptoms, and hemophagocytic syndrome, early bone marrow transplantation is essential in CHS. Griscelli syndrome (GS) also presents with similar silver‐gray hair due to defective melanosome transport caused by pathological variants of MYO5A (GS1), RAB27A (GS2), or MLPH (GS3). However, unlike classic OCA, in which melanin biosynthesis is impaired, GS affects the intracellular transport of melanosomes, leading some to classify it separately from true albinism [30].
Piebaldism
2.4
Piebaldism is an autosomal dominant disorder characterized by partially depigmented patches due to abnormal migration or survival of melanocyte precursor cells and melanoblasts during embryonic development. It is caused by pathogenic variants in KIT, which encodes a receptor tyrosine kinase. It is characterized by a triangular white forelock in the central forehead region, present from birth. Depending on the severity, various degrees of depigmented patches and café‐au‐lait macules are observed on the trunk and limbs. No systemic complications are observed. The severity of depigmented patches and café‐au‐lait macules is primarily determined by the location and type of genetic variants. KIT receptor forms dimers that bind to ligands. When pathogenic variants result in abnormal receptors inhibiting dimer formation, this leads to 50% functional loss (haploinsufficiency), resulting in relatively mild symptoms. Conversely, when dimer formation is preserved but signal transduction is impaired, abnormal receptors also inhibit the function of wild‐type receptors (dominant negative effect), causing 75% functional loss and a tendency toward severe manifestations [31]. However, severity can vary even within families carrying the same variant [32], suggesting the influence of other modifier genes or epigenetic factors.
WS
2.5
WS affects not only melanocytes but also the differentiation and migration of other neural crest‐derived cells, presenting with other organ symptoms in addition to the skin manifestations of piebaldism described above. It is most commonly autosomal dominant, with a global prevalence of approximately 1 in 42 000 individuals. The three cardinal features of this syndrome are (1) pigmentary abnormalities (piebaldism, white forelock, and iris heterochromia), (2) sensorineural hearing loss, and (3) facial characteristics (dystopia canthorum and broad nasal root). A distinctive feature of WS is that its subtypes are classified based on the clinical symptoms rather than the causative genes. In total, WS has four subtypes (WS1–4), which are further subdivided according to the causative genes (Table 2). Special subtypes include the Peripheral demyelinating neuropathy, Central demyelination, WS, and Hirschsprung disease (PCWH) and Tietz albinism‐deafness syndrome caused by the dominant‐negative variants of SOX10 and MITF, respectively [33, 34]. These represent severe forms of WS4 and WS2, with Tietz albinism‐deafness syndrome characterized by generalized hypopigmentation and profound sensorineural hearing loss (Figure 3) [34].
Clinical features of a patient with the Tietz albinism‐deafness syndrome. (a and b) A 3‐year‐old girl presents with blond hair, gray irises, light skin with café‐au‐lait‐like pigmentation, and sensorineural hearing impairment. (c) Fundoscopy revealed hypopigmentation and foveal hypoplasia (left eye). She harbors a dominant negative pathogenic variant in MITF: NM_000248:C.637G>C (p.Glu213Gln).
Hyperpigmentary Disorders
3
DSH
3.1
DSH is a rare pigmentary genodermatosis inherited via autosomal dominant transmission with high penetrance. This condition is predominantly observed in East Asian countries, particularly Japan, China, and Taiwan, with the first case described by Toyama in 1910 [35]. The clinical presentation of DSH is highly characteristic, featuring intermixed hyper‐ and hypopigmented macules on the dorsal regions of the hands and feet, along with freckle‐like macules on the face (Figure 4a–c). This condition usually manifests in infancy or early childhood as faint hypopigmented macules on the dorsal surfaces of extremities. Subsequently, scattered hyperpigmented macules emerge within the hypopigmented regions. Once established, these lesions persist throughout life without progression or regression, and their color and distribution remain essentially unchanged [35].
Clinical and dermoscopic features of patients with dyschromatosis symmetrica hereditaria (DSH; a–c) and lentiginous phenotype due to a SASH1 variant (d–g). (a and b) An 8‐year‐old girl presents with freckle‐like hyperpigmentation on her face and mixed hyper‐ and hypopigmented spots on her dorsal hands and feet. She harbors a heterozygous variant in ADAR: NM_001111.5:C.3200 T>G (p.Leu1067Trp). (c) Dermoscopic image of the dorsal foot of the patient with DSH clearly showing mixed hypo‐ and hyperpigmentation. (d–f) A 38‐year‐old man presents with extensive hyperpigmented macules, predominantly on sun‐exposed areas, which are generally larger than the freckle‐like pigmentation observed in patients with DSH. A heterozygous variant was detected in SASH1: NM_015278.5:C.1930C>T (p.Arg644Trp). (g) Dermoscopic image of the forearm of the patient with the SASH1 variant showing irregularly shaped hyperpigmented macules. Interestingly, perifollicular areas show relative hypopigmentation (arrowheads).
Genetic basis of DSH involves pathogenic variants of adenosine deaminase RNA‐specific (ADAR), which encodes adenosine deaminase acting on RNA1 (ADAR1) [36]. ADAR1 protein catalyzes the conversion of adenosine to inosine in double‐stranded RNA substrates through a process known as A‐to‐I editing. This protein exists as two isoforms: Interferon‐inducible p150 and constitutively expressed p110 forms. The p150 isoform potentially plays an important role in DSH pathogenesis, as some studies have reported DSH development even with pathogenic variants upstream of the p110 initiation codon, which primarily affects p150 expression while preserving p110 function [37]. Although gene penetrance is reportedly 100% in DSH, clinical features may vary among patients with identical variants, suggesting that environmental factors, such as viral infections or ultraviolet exposure, influence phenotypic expression in this disorder.
Histologically, DSH lesions show a markedly lower number of melanocytes in hypopigmented areas than in normal skin. Electron microscopy has revealed degenerative mitochondrial changes and cytoplasmic vacuole formation in melanocytes, indicative of apoptosis [38]. Its pathogenesis likely involves stress‐induced apoptosis of melanocytes harboring ADAR variants, which are potentially triggered by viral infections, leading to the formation of hypopigmented lesions. Subsequently, remnant melanocytes from hair follicle bulge areas possibly migrate to form characteristic hyperpigmented macules.
Notably, ADAR is also a causative gene for the Aicardi–Goutières syndrome (AGS), a severe neuroinflammatory disorder characterized by microcephaly, basal ganglia calcification, and intellectual disability [39]. Compound heterozygous ADAR variants present with overlapping DSH and AGS phenotypes, where patients exhibit both the characteristic skin manifestations of DSH and neurological symptoms of AGS [40]. This phenotypic spectrum suggests that the degree of ADAR1 functional impairment influences the disease severity and manifestations.
DUH
3.2
DUH, first described by Ichikawa and Hiraga in 1933, is characterized by generalized mottled hyperpigmentation and hypopigmentation and distributed throughout the body in a reticulate pattern. This condition typically manifests in infancy or early childhood, presenting with numerous asymptomatic, well‐demarcated, irregular hyperpigmented and hypopigmented macules over the trunk and extremities. DUH is classified into three subtypes: DUH1 (OMIM 127500), DUH2 (OMIM 612715), and DUH3 (OMIM 615402). DUH1 is caused by pathogenic variants in SASH1, a tumor suppressor gene. These variants activate the p53/proopiomelanocortin/α‐melanocyte‐stimulating hormone/Gαs/SASH1 signaling cascade [41], which leads to enhanced phosphorylation of extracellular signal‐regulated kinase‐1/2 and CREB, ultimately resulting in hyperpigmentation [42]. Additionally, SASH1 participates in the Gαs/SASH1/IQ motif‐containing GTPase‐activating protein 1/E‐cadherin pathway that regulates melanocyte transepithelial migration [43]. Importantly, SASH1 variants often present as a distinct clinical entity different from classical DUH. Patients with SASH1 variants frequently exhibit prominent hyperpigmentation, predominantly on sun‐exposed areas, differing significantly from the balanced hyper‐ and hypopigmented patterns (Figure 4d–g) [44]. Due to this predominantly hyperpigmented presentation, SASH1‐related cases are often reported as having a lentiginous phenotype rather than classic DUH [44, 45, 46].
Linkage analysis previously identified 12q21‐q23 as a causative locus for DUH2 [47]. Subsequently, Amyere et al. [48] identified pathogenic variants in KITLG on 12q21.34 in patients with familial progressive hyper‐ and hypopigmentation. Considering the similarity of the clinical manifestations of DUH and FPHH, KITLG is potentially the causative gene for DUH2.
DUH3 is a classic type of DUH characterized by asymptomatic hyperpigmented and hypopigmented macules showing a generalized distribution over the trunk, limbs, and sometimes the face. In 2013, Zhang et al. identified ATP‐binding cassette subfamily B member 6 gene (ABCB6) as the causative gene for this condition [49]. ABCB6 encodes an ATP‐binding cassette transporter that localizes to the lysosomal and early melanosomal membranes. Two major pathogenic mechanisms of DUH have been identified to date. First, pathogenic variants of ABCB6 cause altered subcellular localization, with mutant proteins being retained in the Golgi apparatus, rather than being properly trafficked to melanosomes and dendrites [49]. This disrupts melanosome transport and distribution to keratinocytes. Second, ABCB6 variants impair the early steps of melanogenesis by disrupting premelanosome protein amyloid fibril formation in stage II melanosomes [50]. Premelanosome protein fibrils serve as the structural scaffold for eumelanin deposition, and their disruption leads to aberrant protein aggregation and defective melanosome maturation. These defects possibly contribute to abnormal melanosome function, potentially affecting melanin synthesis, storage, and distribution, which explains the characteristic mixed hyperpigmented and hypopigmented macules observed in patients with DUH. Recently, variants in the period circadian regulator 3 gene (PER3), particularly the PER3 rs772027021 single‐nucleotide variant, have been reported to be associated with the DUH phenotype [51]. This variant may represent a novel DUH subtype, potentially designated as DUH4, characterized by milder pigmentation abnormalities [52].
RAK
3.3
RAK is a rare autosomal dominant pigmentary disorder caused by variants in ADAM10, which encodes a zinc metalloprotease involved in ectodomain shedding of various membrane proteins [53]. First described by Kitamura in 1943, RAK typically manifests in the first or second decade of life with sharply demarcated, reticulate, and slightly depressed brown macules primarily affecting the dorsa of hands and feet (Figure 5) [54]. The macules gradually darken and extend proximally, progressing until middle age before disappearing in the 70s. Additional features include breaks in the epidermal ridges of palms and fingers, palmoplantar pits, and occasional plantar keratoderma and partial alopecia [55].
Clinical features of a patient with reticulate acropigmentation of Kitamura (RAK). (a and b) Reticulate hyperpigmentation on the dorsum of the extremities. (c) Dermoscopic image of hyperpigmented spots showing irregularly shaped hyperpigmentation, without any hypopigmentation. A heterozygous variant was detected in ADAM10:NM_001110.4: C.1931G>C (p.Arg644Pro).
Histologically, RAK exhibits epidermal thinning with elongated and thin rete ridges, showing pigmentation at their pointed ends. The stratum corneum exhibits slight hyperkeratosis without parakeratosis, whereas the superficial dermis shows minimal inflammatory infiltration without pigmentary incontinence. Epidermal melanocyte numbers are increased in this disorder, with electron microscopy showing aggregated melanosomes in melanocytes and an increased number of melanosome complexes in keratinocytes [54].
DDD
3.4
DDD is an autosomal dominant genodermatosis caused by variants of multiple genes, including KRT5, POFUT1, POGLUT1, or PSENEN [56]. The condition presents with reticulate brown macules similar to RAK but demonstrates distinct clinical and temporal characteristics. DDD typically develops around or after the age of 20 years, primarily affecting the flexural areas and major skin folds, rather than the acral regions. In contrast to the uniform light brown macules of RAK, the pigmented lesions appear brown to black in this condition. The key distinguishing features of DDD include comedo‐like follicular papules, genital region involvement, and characteristic dyschromatosis (mixed small hyper‐ and hypopigmented macules in the affected areas). Patients often experience pruritus in skin lesions, which is atypical of RAK. Histologically, DDD shows acanthosis with tight digitiform rete ridges and notable pigmentary incontinence in the dermis, which is in sharp contrast to the epidermal thinning pattern observed in RAK [57].
Both RAK and DDD disrupt the Notch signaling pathway, which plays an important role in melanocyte homeostasis. ADAM10 functions in the ectodomain shedding of Notch proteins as substrates in the skin, whereas POFUT1 and POGLUT1 are essential components involved in the post‐translational modification of Notch proteins via fucosylation and glucosylation, respectively [55]. The gene product of PSENEN also participates in the Notch signaling pathway and is associated with DDD, particularly in cases with concurrent hidradenitis suppurativa [58, 59]. Some case reports have documented patients with DDD with hidradenitis suppurativa harboring pathogenic variants in NCSTN or KRT14 [60, 61]. Impaired Notch signaling disrupts melanocyte homeostasis; however, the precise mechanisms linking Notch pathway dysfunction to the characteristic hyperpigmentation patterns in this condition remain unclear, warranting further research.
RASopathies and Pigmentary Manifestations
3.5
RASopathies are developmental disorders affecting approximately 1 in 1000 individuals that are caused by pathogenic variants in genes encoding components of the RAS–MAPK signaling pathway, which plays a crucial role in melanocyte biology [62, 63]. The RAS pathway regulates essential cellular processes, including growth, differentiation, apoptosis, migration, and senescence. Pathogenic variants of RAS‐related genes result in the dysregulation of these signaling cascades, leading to the characteristic developmental abnormalities and pigmentary changes observed in these conditions. Café‐au‐lait macules are the most common pigmentary abnormalities associated with RASopathies. In neurofibromatosis type 1, loss of neurofibromin disrupts the RAS–GTPase‐activating protein function, resulting in constitutive activation of the RAS–MAPK pathway in melanocytes. Similar café‐au‐lait macules also occur in Legius syndrome, which is caused by pathogenic variants in SPRED1, reflecting its functional convergence with RAS–MAPK signaling [64]. In contrast, McCune–Albright syndrome is caused by the postzygotic activating variants of GNAS, resulting in mosaicism that manifests as large segmental café‐au‐lait macules with jagged “coast of Maine” borders. These lesions follow Blaschko's lines, reflecting the developmental lineage of the affected melanocyte precursors. These pigmentary changes often represent the earliest clinical signs of the McCune–Albright syndrome [65].
Noonan syndrome and related conditions, including cardiofaciocutaneous syndrome and Costello syndrome, present with various pigmentary abnormalities, including café‐au‐lait macules, lentigines, and distinctive hair anomalies. Pathogenic variants in PTPN11 are associated with the Noonan syndrome with multiple lentigines, formerly known as LEOPARD syndrome, which is an acronym for Lentigines, Electrocardiographic abnormalities, Ocular hypertelorism, Pulmonary stenosis, Abnormal genitalia, Retardation of growth, and Deafness [64]. Multiple facial lentigines, along with characteristic facial dysmorphism, represent the key diagnostic features of this condition (Figure 6). The therapeutic implications of understanding these mechanisms have become increasingly apparent with the success of MEK inhibitors, such as selumetinib, in treating RASopathy complications [66], directly demonstrating the ways in which molecular insights translate into clinical advances and enhance our understanding of pigmentation regulation via developmental signaling pathways.
Clinical features of a patient with Noonan syndrome with multiple lentigines. (a) A 6‐year‐old girl presents with multiple lentigines on her face, along with ocular hypertelorism, broad nasal root, and low‐set ears. (b) Café‐au‐lait macules are observed on the patient's body. She harbors a heterozygous variant in PTPN11: NM_002834.5:C.1403C>T (p.Thr468Met).
Future Directions and Conclusion
4
Advances in sequencing technologies have facilitated the identification of novel genes associated with GPDs, enhancing our understanding of the underlying molecular mechanisms and normal pigmentation biology. This increasing genetic knowledge allows the development of precision medicine approaches via improved genotype–phenotype correlations and personalized management strategies based on individual genetic profiles.
GPDs represent a diverse group of genetic conditions that provide important insights into melanocyte biology and pigmentation regulation. Their clinical management requires multidisciplinary approaches addressing not only pigmentary manifestations but also associated complications, such as visual impairment, hearing loss, and cancer predisposition. Early diagnosis and appropriate supportive care remain fundamental for improving the quality of life of the affected individuals. Continued research into these genetic conditions is essential not only for improving patient outcomes but also for advancing our understanding of fundamental cellular mechanisms that extend well beyond melanocyte function, with implications for broader biological processes and therapeutic development.
Ethics Statement
Written informed consent was obtained from all patients or their legal guardians for publication of clinical photographs presented in this review (Figures 1, 2, 3, 4, 5, 6), under protocols approved by the Ethics Committee of the Faculty of Medicine, Yamagata University. All images were modified to protect patient privacy.
Conflicts of Interest
Tamio Suzuki is an editorial board member of the journal of Dermatology and a co‐author of this article. To minimize bias, he was excluded from all editorial decision‐making related to the acceptance of this article for publication.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Y. Yamaguchi and V. J. Hearing , “Physiological Factors That Regulate Skin Pigmentation,” Bio Factors 35 (2009): 193–199.19449448 10.1002/biof.29PMC 2793097 · doi ↗ · pubmed ↗
- 2G. E. Costin and V. J. Hearing , “Human Skin Pigmentation: Melanocytes Modulate Skin Color in Response to Stress,” FASEB Journal 21 (2007): 976–994.17242160 10.1096/fj.06-6649 rev · doi ↗ · pubmed ↗
- 3G. Raposo and M. S. Marks , “Melanosomes–Dark Organelles Enlighten Endosomal Membrane Transport,” Nature Reviews Molecular Cell Biology 8 (2007): 786–797.17878918 10.1038/nrm 2258 PMC 2786984 · doi ↗ · pubmed ↗
- 4A. Kawakami and D. E. Fisher , “The Master Role of Microphthalmia‐Associated Transcription Factor in Melanocyte and Melanoma Biology,” Laboratory Investigation 97 (2017): 649–656.28263292 10.1038/labinvest.2017.9 · doi ↗ · pubmed ↗
- 5Y. Tomita and T. Suzuki , “Genetics of Pigmentary Disorders,” American Journal of Medical Genetics. Part C, Seminars in Medical Genetics 131C (2004): 75–81.15452859 10.1002/ajmg.c.30036 · doi ↗ · pubmed ↗
- 6K. Okamura and T. Suzuki , “Current Landscape of Oculocutaneous Albinism in Japan,” Pigment Cell & Melanoma Research 34 (2021): 190–203.32969595 10.1111/pcmr.12927 · doi ↗ · pubmed ↗
- 7A. B. Murthy , V. Palaniappan , K. Karthikeyan , and V. Anbarasan , “Dyschromatosis Universalis Hereditaria,” International Journal of Dermatology 62 (2023): 1218–1227.37634201 10.1111/ijd.16817 · doi ↗ · pubmed ↗
- 8L. Mohapatra , K. Sardana , M. Panda , and R. Mahajan , “An Algorithmic Approach Towards Diagnosis of Patients With Hereditary Reticulate Pigmentary Disorders: A Narrative Review,” Clinical and Experimental Dermatology 50 (2024): 12–20.39139099 10.1093/ced/llae 322 · doi ↗ · pubmed ↗