Mutant knock-in mice display enhanced susceptibility to pure prion protein fibrils
Daniel J. Walsh, Heidi Standke, Allison Kraus, Joel C. Watts, Surachai Supattapone

TL;DR
Mice with a specific prion protein mutation are more susceptible to prion disease caused by pure prion protein fibrils.
Contribution
The study reveals that the E200K mutation increases host susceptibility to protein-only prion fibrils.
Findings
Protein-only PrPSc molecules induce PrPSc formation and brain degeneration in E200K knock-in mice.
These effects do not occur in mice expressing wild-type prion protein.
The E200K mutation suggests differences in replication mechanisms between mutant and wild-type prions.
Abstract
Prion diseases manifest clinically in three different forms. Sporadic and infectious forms of prion disease are caused by the conversion of WT, cellular prion protein (PrPC) into its pathogenic conformer (PrPSc). In contrast, genetic forms of prion diseases are caused by mutations in the PrP sequence that promote mutant PrPSc formation. When reconstituted with either polyanionic or lipid cofactors, purified PrPC substrate can be converted in vitro into PrPSc products that display high levels of specific infectivity when inoculated in WT hosts. In contrast, various protein-only PrPSc molecules formed in the absence of cofactors display much lower levels of specific infectivity. Here, we report that protein-only PrPSc molecules with different sequences can induce the formation of proteinase K-resistant PrPSc molecules and spongiform degeneration in the brains of knock-in mice expressing…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
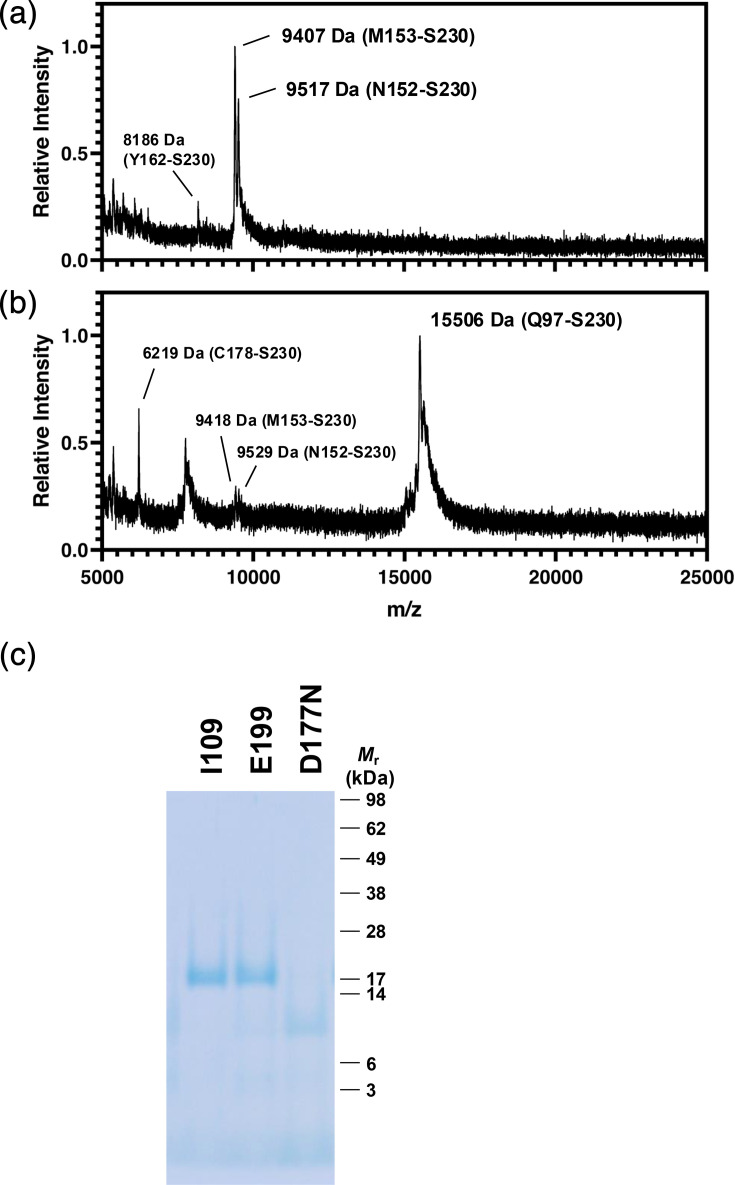 Fig. 1
Fig. 1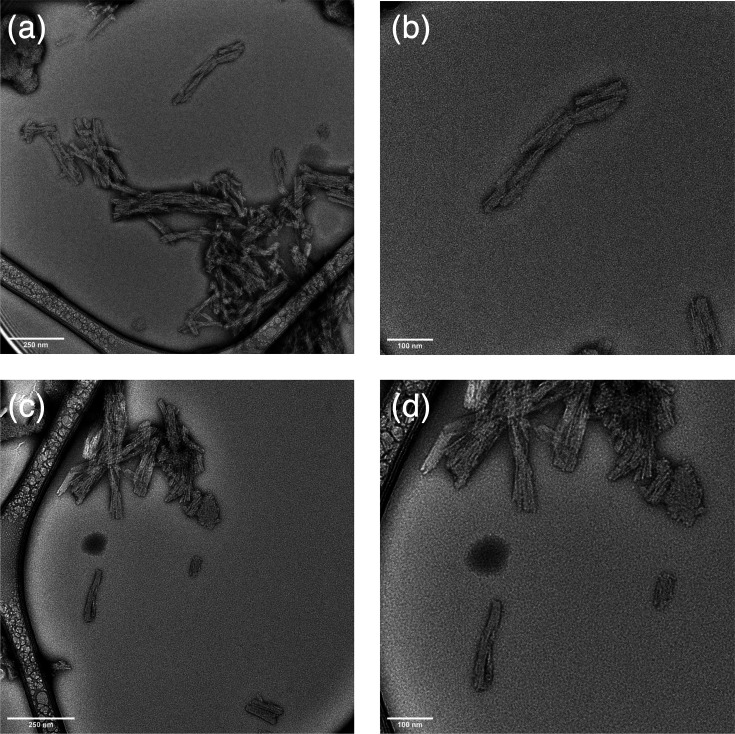 Fig. 2
Fig. 2 Fig. 3
Fig. 3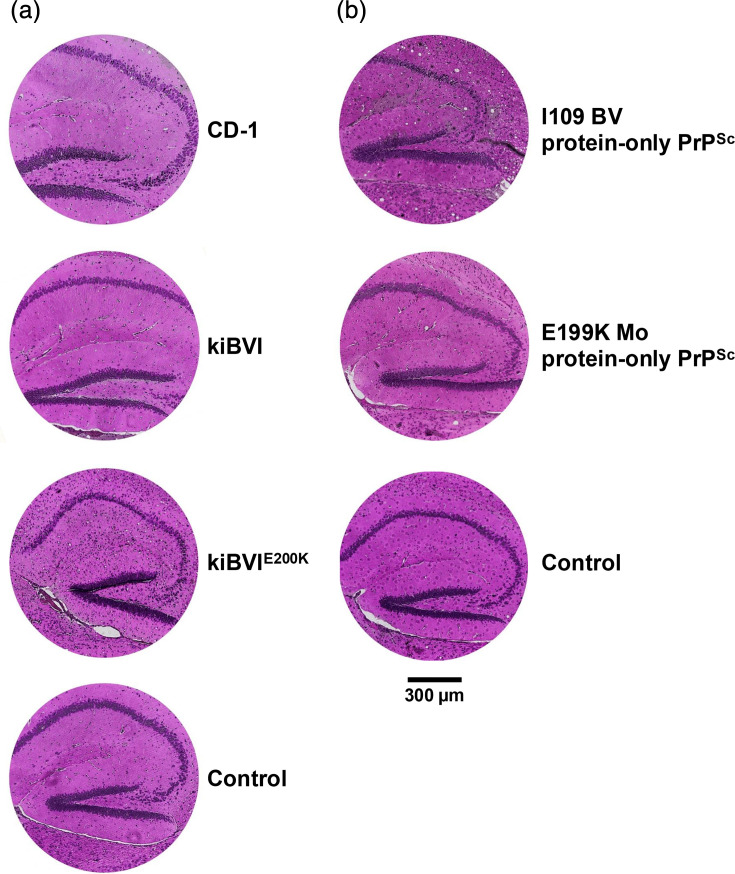 Fig. 4
Fig. 4| CD-1 mice | Negative control | 0/8 | >700 |
| E199K Mo protein-only PrPSc | 0/4 | >700 | |
| D177N Mo protein-only PrPSc | 0/4 | >700 | |
| M109 bank voles | Negative control | 0/8 | >700 |
| I109 BV protein-only PrPSc | 0/3 | >700 | |
| E199K Mo protein-only PrPSc | 0/4 | >700 | |
| D177N Mo protein-only PrPSc | 0/7 | >700 | |
| kiBVI mice | Negative control | 0/5 | >650 |
| I109 BV protein-only PrPSc | 0/4 | >650 | |
| E199K Mo protein-only PrPSc | 0/7 | >600 | |
| D177N Mo protein-only PrPSc | 0/10 | >600 |
| kiBVID178N mice | Negative control | 7/7 | 518+21 |
| I109 BV protein-only PrPSc | 4/4 | 466+85 | |
| E199K Mo protein-only PrPSc | 6/6 | 485+26 | |
| D177N Mo protein-only PrPSc | 6/6 | 499+8 | |
| kiBVIE200K mice | Negative control | 8/8 | 646+70 |
| I109 BV protein-only PrPSc | 3/3 | 625+37 | |
| E199K Mo protein-only PrPSc | 5/5 | 603+46 | |
| D177N Mo protein-only PrPSc | 9/9 | 336+23† |
- —http://dx.doi.org/10.13039/100000065 National Institute of Neurological Disorders and Stroke
- —http://dx.doi.org/10.13039/100000065 National Institute of Neurological Disorders and Stroke
- —http://dx.doi.org/10.13039/100000065 National Institute of Neurological Disorders and Stroke
- —http://dx.doi.org/10.13039/100000057 National Institute of General Medical Sciences
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPrion Diseases and Protein Misfolding · Bacteriophages and microbial interactions · HIV Research and Treatment
Introduction
Prion diseases are fatal neurodegenerative disorders caused by the misfolding of the host-encoded prion protein [cellular prion protein (PrP^C^)] into an aggregated conformer (PrP^Sc^) [12]. Human cases fall into three broad categories – genetic, sporadic and infectious – which differ in origin, epidemiology and clinical features [3]. Genetic forms of disease such as familial Creutzfeldt–Jakob disease (fCJD), Gerstmann–Sträussler–Scheinker and fatal familial insomnia (FFI) are caused by pathogenic PrP mutations such as E200K in fCJD and D178N in FFI [45]. These mutations drive the formation of mutant PrP^Sc^ molecules and subsequent neurodegeneration in human patients. In contrast, sporadic and infectious forms of prion disease, such as sporadic CJD, scrapie, bovine spongiform encephalopathy and chronic wasting disease are caused by the auto-catalytic conversion of WT PrP^C^ into PrP^Sc^ by an unknown mechanism.
Our lab and others have replicated various synthetic PrP^Sc^ molecules (operationally defined here as protease-resistant PrP conformers that self-propagate faithfully and efficiently) in vitro with markedly different levels of specific infectivity. Collectively, these studies have shown that non-proteinaceous cofactors are required to produce WT prions with high levels of specific infectivity [611]. Interestingly, in the absence of cofactor molecules, both WT and mutant PrP substrates can also form self-replicating amyloid fibrils with PIRIBS (parallel in-register intermolecular β-sheet) architecture, but these ‘protein-only’ fibrils display relatively low levels of specific infectivity in hosts expressing WT PrP^C^ molecules without amyloidogenic polymorphisms or mutations, such as I109, D178N or E200K [7,1115]. This is true even for protein-only PrP^Sc^ molecules seeded with the same template as fully infectious cofactor PrP^Sc^ molecules [7].
Knock-in (ki) mice expressing bank vole (BV) PrP^C^ molecules with the I109 polymorphism (kiBVI) have recently been developed [16]. The I109 polymorphism promotes the spontaneous formation of PrP^Sc^ in transgenic mice overexpressing I109 BV PrP [17], but kiBVI mice do not develop spontaneous disease. In contrast, ki mice expressing I109 BV PrP containing D178N and E200K mutations (kiBVI^D178N^ and kiBVI^E200K^, respectively) spontaneously develop prion disease associated with spongiform degeneration [16]. However, spontaneously sick kiBVI^D178N^ and kiBVI^E200K^ mice do not appear to accumulate classical forms of proteinase K (PK)-resistant PrP^Sc^ in their brains [16]. Because prior studies suggest that different mechanisms may be responsible for the propagation of WT and mutant prions (i.e. with PrP sequences containing pathogenic mutations such as D178N or E200K or amyloidogenic polymorphisms such as I109) [1820], we sought to determine whether any of these new ki mouse lines might be more susceptible to various protein-only PrP^Sc^ molecules than WT hosts.
Methods
In vitro propagation of protein recPrPSc samples
Cocktails for protein-only D177N and E199K mouse (Mo) PrP^Sc^ serial propagation reactions were prepared as previously described [18]. Unseeded reactions containing 6 µg ml^−1^ purified Mo recPrP 23–230 (either D177N or E199K) in conversion buffer (20 mM Tris, 135 mM NaCl, 5 mM EDTA pH 7.5, 0.15% (v/v) Triton X-100, pH 7.4) without cofactors in a total volume of 400 µl were shaken in 1.5 ml microfuge tubes and serially propagated at a 20% (v/v) seeding ratio. Shaking was performed as previously described [19] in a home-built machine with an 8 mm orbit shaking at ∼1,200 r.p.m. Each reaction was shaken for 72 h at a temperature of 37 °C before propagation to the next round.
Cocktails for protein-only I109 BV serial propagation reactions containing 6 µg ml^−1^ purified I109 BV recPrP 23–230 in conversion buffer without cofactors were prepared and initially seeded with protein-only I109 BV protein-only PrP^Sc^ generated by protein misfolding cyclic amplification (PMCA) as previously described [11] and similarly shaken and serially propagated at 1,200 r.p.m. in our custom-built shaker, as described above for mutant PrP samples.
Rodent models
CD-1 mice were purchased from Charles River Laboratories (Worcester, MA). BVs were kindly provided by Umberto Agrimi (Rome, Italy). The ki mouse models kiBVI (expressing I109 BV PrP), kiBVI^D178N^ (expressing I109 D178N BV PrP) and kiBVI^E200K^ (expressing I109 E200k BV PrP) used in this study have been previously described [16].
Inoculation, diagnosis and tissue harvest
Intracerebral inoculation and diagnosis of prion disease were performed as described [1121] with the following modifications. In vitro-converted mutant PrP^Sc^ samples were dispersed by three 20 s sonication pulses in a cup horn sonicator (Qsonica Misonix, Newtown, CT) and subsequently diluted tenfold into PBS+1% BSA to be used as inoculum (i.e. final concentration of PrP^Sc^=0.6 µg ml^−1^). The inoculum volume used was 30 µl. Rodents were inoculated between 4 and 6 weeks of age or allowed to age without intervention until the onset of scrapie. At terminal disease stage, mice were euthanized and brains were recovered. Brains for biochemical analysis were immediately frozen at −70 °C, and brains for histopathology were fixed in PBS+4% formalin.
Proteinase K digestion of PrPSc
Brain homogenates (10% (w/v) in PBS) from experimental mouse brains were digested in a reaction containing 10 µg ml^−1^ PK, 1% (v/v) Triton X-100, 0.5% sodium deoxycholate at 37 °C with shaking at 1000 r.p.m. for 1 h. PrP^Sc^ samples produced in vitro were treated with 20 µg ml^−1^ PK at 37 °C for 30 min. All digestion reactions were quenched by addition of 4 mM PMSF.
ReadyBlue-stained SDS-PAGE
Aliquots of recombinant protein-only PrP^Sc^ samples produced in vitro were treated with 20 µg ml^−1^ PK at 37 °C for 30 min. All digestion reactions were quenched by the addition of 4 mM PMSF. Samples were centrifuged at 18,000 g for 1 h at 4 °C. Supernatants were discarded, and pellets were resuspended in ~60 µl total volume Lauryl dodecyl sulfate sample buffer (Invitrogen, Carlsbad, CA). All samples were boiled for 7 min, and 25 µl of each sample was loaded into a pre-cast 10% NuPage Bis-Tris gel (Invitrogen) alongside a molecular weight marker lane containing SeeBlue Plus 2 prestained standard (Invitrogen) and run in MES SDS running buffer (Invitrogen) at 200V for 35 min. The gel was subsequently stained with ReadyBlue Protein Gel Stain (Sigma Aldrich, St. Louis, MO) and imaged with an Azure 600 imager (Dublin, CA).
Western blotting
All protein samples were denatured by boiling at 95 °C in Laemmli SDS sample buffer (Bioland Scientific) for 15 min. Denatured protein samples were run on 12% SDS-PAGE gels and transferred to an Immobilon-P PVDF membrane (Millipore, Burlington, MA) using a semi-dry blotting apparatus (Bio-Rad, Hercules, CA). Western blots were probed using anti-PrP D18 primary antibody (epitope 132–156)and HRP-linked goat anti-human secondary antibody (Invitrogen).
Histopathology
Neuropathology was performed as previously described [22]. Formalin-fixed brains were disinfected by immersion in 96% formic acid for 1 h. Tissue blocks and 4 µm thick microscopic sections stained with haematoxylin and eosin were prepared by the Dartmouth Hitchcock Research Pathology Service Core (Lebanon, NH), and vacuolation in various brain regions was scored on a 0–5 scale.
MALDI-TOF mass spectrometry
To prepare mutant Mo PrP^Sc^ conformers for mass analysis, 5 ml of converted cocktail was first treated with PK, as described above, and then quenched with PMSF. N-Octyl-β-d-glucopyranoside (Anatrace, Maumee, OH) detergent was added to a final concentration of 1% (w/v), and samples were end-over-end rotated for 30 min at room temperature to allow for full solubilization. After 1 h of centrifugation at 18,000 g, the supernatant was discarded, and pellets were resuspended in an equal volume of water. This centrifugation and water wash step was then repeated to remove any residual buffer components. After a third centrifugation, pellets were denatured by resuspending in 100 µl of 6 M guanidine hydrochloride and incubating for 3 h at 60 °C, shaking at 900 r.p.m. After centrifugation again at 18,000 g for 1 h, denatured protein was isolated from the supernatants by methanol/chloroform precipitation [23]. Protein pellets from the interphase were dried and then resuspended in 30 µl water.
MALDI-TOF mass spectrometry was performed as described previously [24]. Briefly, the protein sample was concentrated using an OMIX C4 chromatographic pipette tip (Varian Inc., Santa Clara, CA) and eluted with sinapinic acid matrix onto a Voyager 100-position MALDI sample plate (Applied Biosystems, Foster City, CA). Spectra were acquired using a Voyager-DE Pro Biospectrometry Workstation (Applied Biosystems) with a mass accuracy of ±10 Da.
Electron microscopy
To prepare D177N recPrP^Sc^ for negative stain imaging, n-octyl-β-d-glucopyranoside was added to shaken cocktails at 1% (w/v) to solubilize unconverted protein, and solutions were centrifuged at 4,000 g to collect fibrillar material. Pellets were subjected to two additional centrifugation and wash steps with 1/3× PBS, 0.02% amphipol to remove detergent and concentrate the sample for application to electron microscopy grids.
Four hundred mesh lacey carbon grids (Ted Pella, Redding, CA) were glow discharged and placed on a 7 µl droplet of concentrated D178N PrP^Sc^ sample and incubated for 20 min in a humidified chamber. Grids were then blotted on filter paper, washed in Nano-W stain (Nanoprobes, Yaphank, NY), blotted again and then stained by placing on a droplet of Nano-W for 1 min. Grids were then blotted dry and imaged on an FEI (Field Electron and Ion Company) Tecnai TF20 (200 kV, FEG) with a 4k × 4k CMOS (Complementary Metal-Oxide-Semiconductor)-based Tietz TemCam-F416 camera.
RESULTS
We first inoculated three different preparations of protein-only PrP^Sc^ molecules into WT hosts and kiBVI mice. I109 BV protein-only PrP^Sc^, D177N Mo protein-only PrP^Sc^ and E199K Mo protein-only PrP^Sc^ (note D177N and E199K in Mo PrP correspond to D178N and E200K BV and human PrP) were generated as previously described [1118] and propagated by shaking in the absence of cofactor molecules [19]. CD-1 mice, WT BVs and kiBVI mice all remained healthy for >600 days following inoculation with any of the three protein-only PrP^Sc^ preparations (Table 1). Next, we inoculated the same three protein-only PrP^Sc^ preparations into kiBVI^D178N^ and kiBVI^E200K^ mice. As expected, uninoculated kiBVI^D178N^ and kiBVI^E200K^ control mice developed spontaneous disease (Table 2). Inoculation of various protein-only PrP^Sc^ molecules did not cause statistically significant changes in disease-free survival in either line of ki mice, with one exception: inoculation of D177N Mo protein-only PrP^Sc^ into kiBVI^E200K^ mice reduced disease-free survival from ~650 days to ~340 days (P-value=0.0003) (Table 2).
Interestingly, the PK-resistant core of D177N Mo protein-only PrP^Sc^ is much smaller than that of other cofactor and protein-only PrP^Sc^ conformers [71114151819]. Using MALDI-TOF mass spectrometry, we determined that the PK-resistant core of D177N Mo protein-only PrP^Sc^ encompasses residues 152–230 (Fig. 1a); for comparison, the PK-resistant core of E199K Mo protein-only PrP^Sc^ is 97–230 (Fig. 1b). It should be noted that both E199K and D177N PrP^Sc^ molecules can generate both PK-resistant core sizes. In serial PMCA reactions, the relative amounts of the two sizes are similar [18], whereas in continuous shaking reactions, the relative distributions skew towards E199K PrP^Sc^ having predominantly the larger core and D177N PrP^Sc^ having predominantly the smaller core, but both sizes of PK-resistant bands remain visible for both conformers by SDS-PAGE (Fig. 1c) and Western blot [19]. Despite the relatively small size of its predominant protease-resistant core, D177N Mo protein-only PrP^Sc^ forms amyloid fibrils similar to a variety of other PrP^Sc^ conformers with larger protease-resistant cores [15,2531], as determined by negative-stain electron microscopy (Fig. 2).
Mass analysis of protease-resistant domains in mutant PrPSc conformers. MALDI-TOF mass spectra of (a) D177N and (b) E199K Mo PrPSc conformers after digestion with proteinase K. Peaks unambiguously assigned are labelled with both the mass and the identity of the fragment. Unlabelled peaks either could not be confidently assigned or represent the z=2 charge state of the majority fragment. (c) ReadyBlue-stained SDS-PAGE of I109 BV, E199K Mo and D177N Mo protein-only PrPSc samples following limited proteolysis with proteinase K.
D177N recPrPSc assembles into fibrillar aggregates. Negative stain electron micrographs of shaken D177N recPrPSc fibrils at varying magnification. Scale bars are 250 nm (a, c) and 100 nm (b, d).
To test whether protein-only PrP^Sc^ inocula induced formation of new PrP^Sc^ molecules in the brains of host animals, we digested experimental brain homogenates with PK and visualized PK-resistant PrP^Sc^ molecules with Western blotting (?Fig. 3). The results show that both D177N Mo protein-only PrP^Sc^ and I109 BV protein-only PrP^Sc^ induced formation of PK-resistant PrP^Sc^ in the brains of kiBVI^E200K^ mice, but not in WT mice or voles, kiBVI mice or kiBVI^D178N^ mice (?Fig. 3a). PK-resistant PrP^Sc^ could also be detected in the brains of some but not all kiBVI^E200K^ mice inoculated with E199K Mo protein-only PrP^Sc^ (?Fig. 3b). Taken together, these results suggest that kiBVI^E200K^ mice are a relatively good host for protein-only PrP^Sc^ inocula.
Western blots of proteinase K-resistant PrPSc molecules in the brains of experimental animals. (a) Brain homogenates prepared from various rodent hosts inoculated with either D177N Mo protein-only PrPSc or I109 BV protein-only PrPSc, as indicated. PK=proteinase K digestion. (b) Brain homogenates prepared from kiBVIE200K mice inoculated with various protein-only PrPSc preparations, as indicated. Samples taken from two independent animals are shown for each inoculum. -PK=sample not digested with proteinase K.
It is known that kiBVI^E200K^ mice spontaneously develop scrapie clinical signs with spongiform vacuolation at ~20 months of age [1620]. To evaluate whether protein-only PrP^Sc^ inocula induce any additional neuropathological changes in kiBVI^E200K^ hosts, we assessed vacuolation specifically within the hippocampus, a brain region that displays only minimum levels of vacuolation in spontaneously sick kiBVI^E200K^ mice [1620]. The results show that D177N protein-only PrP^Sc^ molecules induce vacuolation in the hippocampus of kiBVI^E200K^ mice, but not in CD-1 or kiBVI mice (?Fig. 4a, compare the third row to the top two rows). Inoculation of I109 BV protein-only PrP^Sc^ also induces vacuolation in the hippocampus of kiBVI^E200K^ mice (?Fig. 4b, top row), but inoculation of E199K Mo protein-only PrP^Sc^ does not (?Fig. 4b, second row). Overall, our results indicate that kiBVI^E200K^ mice are relatively good hosts for various protein-only PrP^Sc^ inocula, but some protein-only PrP^Sc^ molecules are more efficient than others at inducing the formation of mutant PrP^Sc^ molecules, vacuolation and clinical signs.
Neuropathology of rodents inoculated with protein-only PrPSc inocula. Representative microscopic images of the hippocampus in brain sections stained with haematoxylin and eosin (H and E) (a) Various rodents inoculated with D177N Mo protein-only PrPSc, as indicated. Asymptomatic CD-1 mouse=8 months old, asymptomatic kiBVI mouse=20 months old, symptomatic kiBVIE200K mouse=8 months old. Control=Uninoculated kiBVIE200K mouse (14 months old, asymptomatic). (b) kiBVIE200K mice inoculated with alternative protein-only PrPSc inocula, as indicated. Symptomatic mouse inoculated with I109 BV protein-only PrPSc=23 months old, symptomatic mouse inoculated with E199K Mo protein-only PrPSc=19 months old. Control=Uninoculated kiBVIE200K mouse (23 months old, asymptomatic).
DISCUSSION
In this manuscript, we report that kiBVI^E200K^ mice are far more susceptible to protein-only PrP^Sc^ molecules than either WT hosts or kiBVI mice. Previous in vitro studies showed that cofactor molecules facilitate the formation of WT PrP^Sc^ (even when seeded by mutant prions), whereas mutant PrP substrate molecules can spontaneously convert into PrP^Sc^ without cofactor molecules [1820]. Together with the results of those studies, the in vivo data reported here suggest that WT and mutant PrP^Sc^ molecules replicate by distinct mechanisms.
Western blot data indicate that various protein-only PrP^Sc^ molecules can propagate in kiBVI^E200K^ mice, but not in kiBVI or kiBVI^D178N^ mice. Even though transgenic mice overexpressing I109 BV PrP develop spontaneous prion disease, our results indicate that addition of the pathogenic E200K mutation is important for susceptibility to protein-only PrP^Sc^ inocula. The apparent inability of kiBVI^D178N^ mice to replicate various protein-only PrP^Sc^ molecules, even D177N Mo protein-only PrP^Sc^, is most likely an artefact due to the lower PrP^C^ expression level in kiBVI^D178N^ mice (~60% lower) compared to kiBVI^E200K^ mice [16].
Our data indicate that kiBVI^E200K^ mice were most susceptible to D177N Mo protein-only PrP^Sc^ and least susceptible to E199K Mo protein-only PrP^Sc^. Notably, inoculation with D177N Mo protein-only PrP^Sc^ accelerates the onset of scrapie clinical signs, induces the formation of PK-resistant PrP^Sc^ and causes specific vacuolation in the hippocampus of kiBVI^E200K^ mice. In contrast, I109 BV protein-only PrP^Sc^ induces the formation of PK-resistant PrP^Sc^ and vacuolation but does not accelerate disease, and E200K Mo protein-only PrP^Sc^ induces the formation of PK-resistant PrP^Sc^ without causing either vacuolation or disease acceleration. The structural basis for the relative pathogenicity of various protein-only PrP^Sc^ molecules in kiBVI^E200K^ mice remains unknown, but our results show neither the I109 BV backbone nor the E200K mutation in PrP^Sc^ is necessary for efficient transmission. Surprisingly, the most pathogenic inoculum, D177N Mo protein-only PrP^Sc^, (1) has the lowest degree of homology to the PrP^C^ sequence expressed in kiBVI^E200K^ mice, and (2) has predominantly a much smaller PK-resistant, structured core compared to the other two inocula (in addition to a small amount of a larger PK-resistant core). Typically, prion transmissions are more efficient when there is (1) greater sequence similarity between the inoculated PrP^Sc^ and host PrP^C^ and (2) a larger PK-resistant core [32]. We infer that I109 BV protein-only PrP^Sc^ and E199K Mo protein-only PrP^Sc^ were not able to accelerate clinical disease because they were unable to induce the formation of host PrP^Sc^ as quickly as D177N Mo protein-only PrP^Sc^, relative to the regular lifespan of the host animals. Interestingly, FFI caused by the D178N mutation has much higher clinical penetrance than fCJD caused by the E200K mutation in human patients [33] despite D178N PrP^Sc^ having a smaller PK-resistant core than E200K PrP^Sc^ [34]. Future structural and functional studies of these three protein-only PrP^Sc^ conformers may help us better understand their differences in pathogenicity.
It remains unclear why mutant prions can be formed without cofactors. Structural analysis by X-ray crystallography [35], nuclear magnetic resonance [36] and molecular dynamics simulations [233738] has suggested that disease-associated mutations can disrupt salt bridge and hydrogen bonding networks present in the normal cellular conformation of the protein, PrP^C^, leading to a destabilization of the alpha helix-rich fold and increased propensity for misfolding. It is possible that this destabilization circumvents the role normally played by cofactor molecules during the formation of WT prions.
In summary, we report the first set of in vivo experimental results which support the hypothesis that hosts expressing mutant and polymorphic PrP^C^ molecules use a different mechanism than WT hosts to replicate prions.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Caughey BW Dong A Bhat KS Ernst D Hayes SF et al Secondary structure analysis of the scrapie-associated protein Pr P 27-30 in water by infrared spectroscopy Biochemistry 1991307672768010.1021/bi 00245 a 0031678278 · doi ↗ · pubmed ↗
- 2Pan KM Baldwin M Nguyen J Gasset M Serban A et al Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins Proc Natl Acad Sci U S A 199390109621096610.1073/pnas.90.23.109627902575 PMC 47901 · doi ↗ · pubmed ↗
- 3Prusiner SB Prions Proc Natl Acad Sci U S A 199895133631338310.1073/pnas.95.23.133639811807 PMC 33918 · doi ↗ · pubmed ↗
- 4Chen C Dong XP Epidemiological characteristics of human prion diseases Infect Dis Poverty 201654710.1186/s 40249-016-0143-827251305 PMC 4890484 · doi ↗ · pubmed ↗
- 5Kovács GG Trabattoni G Hainfellner JA Ironside JW Knight RSG et al Mutations of the prion protein gene phenotypic spectrum J Neurol 20022491567158210.1007/s 00415-002-0896-912420099 · doi ↗ · pubmed ↗
- 6Deleault NR Harris BT Rees JR Supattapone S Formation of native prions from minimal components in vitro Proc Natl Acad Sci U S A 20071049741974610.1073/pnas.070266210417535913 PMC 1887554 · doi ↗ · pubmed ↗
- 7Deleault NR Walsh DJ Piro JR Wang F Wang X et al Cofactor molecules maintain infectious conformation and restrict strain properties in purified prions Proc Natl Acad Sci U S A 2012109 E 19384610.1073/pnas.120699910922711839 PMC 3396481 · doi ↗ · pubmed ↗
- 8Katorcha E Gonzalez-Montalban N Makarava N Kovacs GG Baskakov IV Prion replication environment defines the fate of prion strain adaptation P Lo S Pathog 201814 e 100709310.1371/journal.ppat.100709329928047 PMC 6013019 · doi ↗ · pubmed ↗