SmartCrop: knowledge base of molecular genetic mechanisms of rice and wheat adaptation to stress factors
P.S. Demenkov, T.V. Ivanisenko, M.A. Kleshchev, E.A. Antropova, I.V. Yatsyk, A.R. Volyanskaya, A.V. Adamovskaya, A.V. Maltseva, A.S. Venzel, H. Chao, M. Chen, V.A. Ivanisenko

TL;DR
SmartCrop is a knowledge base that helps researchers understand how rice and wheat adapt to environmental stresses at the molecular level.
Contribution
The novel contribution is the development of SmartCrop, an adapted version of the ANDSystem technology for plant research.
Findings
SmartCrop assists in interpreting omics data and reconstructing gene networks in rice and wheat.
It identifies regulatory pathways and candidate genes for stress resistance in crops.
The tool supports marker-assisted selection and discovery of targets for plant protection agents.
Abstract
The study of molecular genetic mechanisms of plant responses to specific growth conditions and stress factors is a central focus of scientific research aimed at developing new valuable crop varieties, particularly rice and wheat. These factors include abiotic stresses (high or low temperatures, drought, salinity, soil metal contamination), biotic stresses (pathogens, pests), as well as plant responses to regulatory factors (fertilizers, hormones, elicitors, and other compounds). Modern research in plant genetics is based on the understanding that the formation of any phenotypic characteristics (molecular genetic, biochemical, physiological, morphological, etc.) is controlled by gene networks – groups of coordinately functioning genes interacting through their products (RNA, proteins, and metabolites). Previously, we developed the ANDSystem intelligent technology designed to extract…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
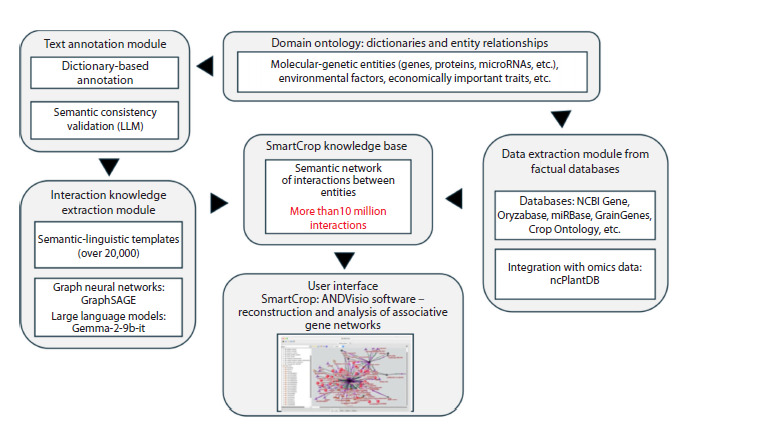 Fig. 1
Fig. 1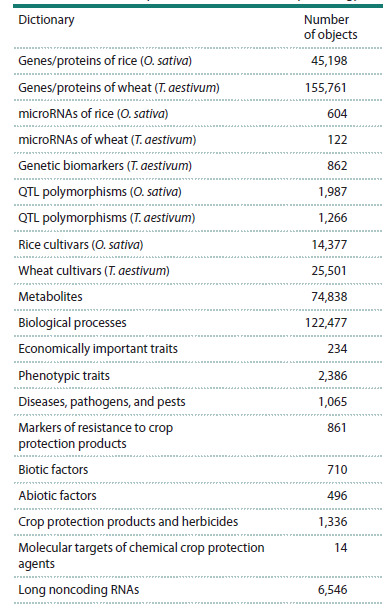 Table 1
Table 1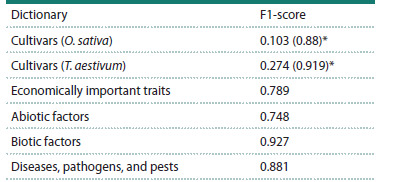 Table 2
Table 2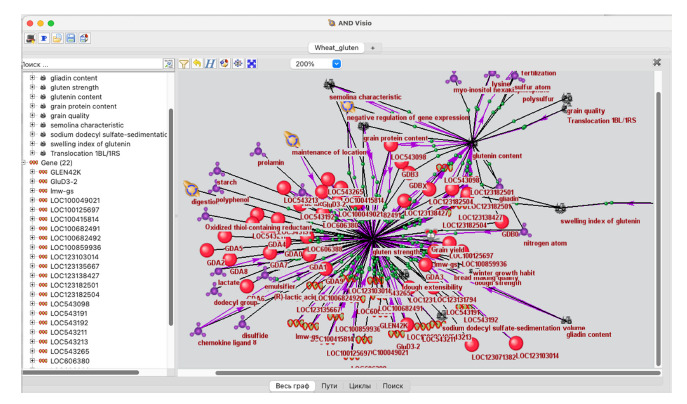 Fig. 2
Fig. 2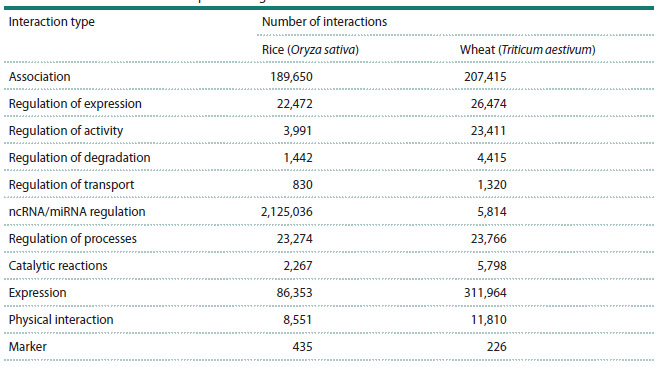 Table 3
Table 3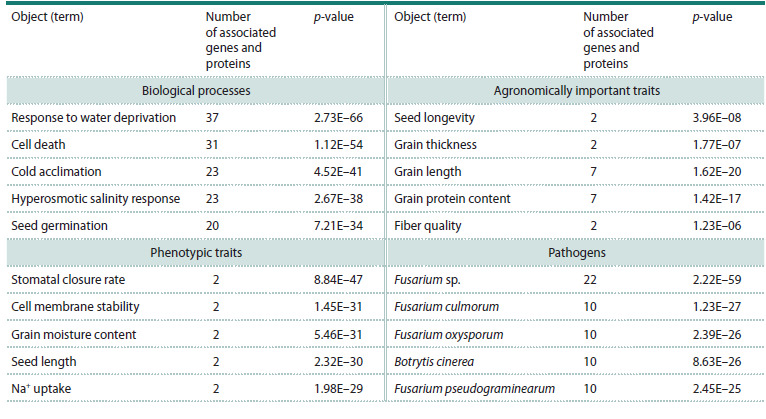 Table 4
Table 4 Fig. 3
Fig. 3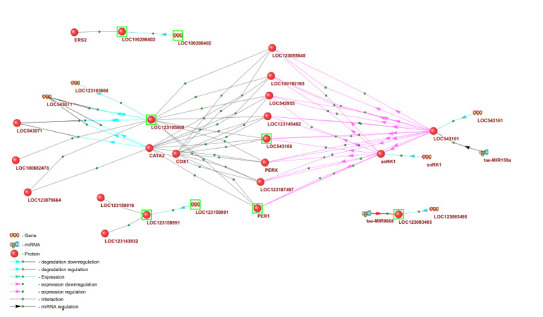 Fig. 4
Fig. 4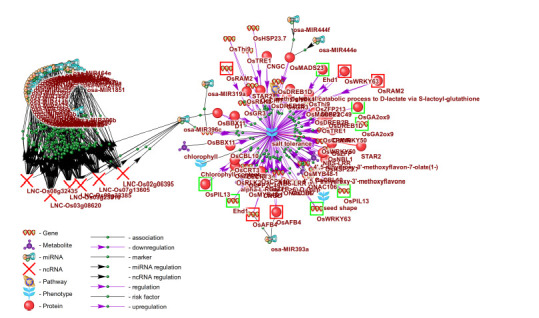 Fig. 5
Fig. 5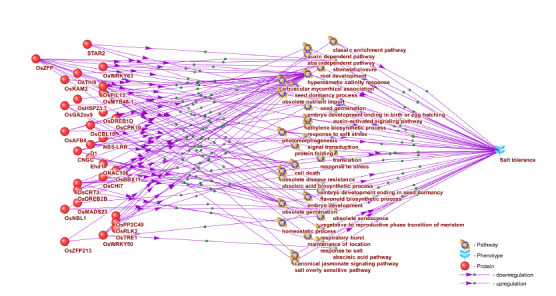 Fig. 6
Fig. 6Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenetics, Bioinformatics, and Biomedical Research · Wheat and Barley Genetics and Pathology · Bioinformatics and Genomic Networks
Introduction
Rice (Oryza sativa L.) and wheat (Triticum aestivum L.) are among the most important agricultural crops, ensuring food security for a significant portion of the world’s population. Both crops are well known for their high nutritional, industrial, and fodder value (Shewry, Hey, 2015). Under current conditions, the production of these crops faces serious challenges. Extreme weather events, adverse climate change, plant diseases, and pests lead to substantial yield losses (Lesk et al., 2016). Overcoming these difficulties is impossible without studying the molecular genetic mechanisms underlying plant resistance to unfavorable biotic and abiotic factors, which requires the analysis of complex systems that include intricate signaling, regulatory, transport, and metabolic pathways (Mittler, 2006; Nykiel et al., 2023).
An effective tool for studying such mechanisms is gene networks, which control molecular genetic processes that determine the formation of phenotypic traits and the functioning of biological processes, including plant stress responses.
The modern concept of gene networks encompasses not only molecular components (RNAs, genes, proteins, and metabolites) but also a wide range of heterogeneous entities, including diseases, biological processes, and environmental factors. This type of gene network is known as an associative gene network (Ivanisenko V.A. et al., 2015). Structurally, such networks represent a knowledge graph integrating information about interactions among diverse objects involved in the functioning of molecular genetic systems or influencing them. In agrobiology and crop science, gene network analysis is successfully used to study economically important traits such as resistance to diseases and pests, tolerance to abiotic stress factors, and yield (Virlouvet et al., 2018; Chen et al., 2020).
The reconstruction of plant gene networks is a complex task that requires processing massive amounts of data and integrating fragmented information from scientific publications, including data on regulatory, transport, and catalytic processes, as well as relationships between genetic features, phenotypic manifestations, and environmental factors. To extract such knowledge, text-mining methods are applied, based both on classical computational approaches (dictionary-based methods, syntactic and linguistic rules and patterns, statistically significant co-occurrence, etc.) and on machine-learning techniques (Ivanisenko T.V. et al., 2014; Shrestha et al., 2024; Zhang et al., 2024).
Machine-learning algorithms used for constructing and analyzing gene networks can be divided into the following categories: supervised learning, unsupervised learning, semi-supervised learning, and hybrid approaches. Supervised learning methods rely on pre-annotated data to build predictive models, for example, to identify key regulators or to predict functional interactions between plant genes (Ni et al., 2016). Unsupervised learning enables the discovery of hidden patterns in large datasets, which is important, for instance, when clustering genes based on expression similarity or identifying gene-network modules. Semi-supervised learning combines the strengths of both approaches, using both labeled and unlabeled data, which is particularly relevant when the amount of well-annotated data is limited (Yan, Wang, 2022). Hybrid approaches integrate various machine-learning methods as well as traditional bioinformatics techniques, allowing them to effectively compensate for the limitations each approach may have when used alone (Guindani et al., 2024; Ivanisenko T.V. et al., 2024). For example, combining dictionary-based named-entity recognition in texts with machine-learning methods improves the accuracy of entity identification (Do et al., 2018; Ivanisenko T.V. et al., 2020).
In recent years, deep machine learning has achieved significant advances through the introduction of the transformer architecture and attention mechanisms, which have enabled substantial progress in natural language processing and the analysis of biological sequences (Vaswani et al., 2017). The analysis of gene networks has also seen considerable development with the application of graph neural networks, among which the GraphSAGE architecture (Hamilton et al., 2017) enables efficient training on large heterogeneous graphs by aggregating features from neighboring nodes. A promising direction is the use of large language models, such as Gemma-2-9b-it (Gemma Team, Google DeepMind, 2024), which provide high-quality semantic analysis of scientific texts and validation of extracted interactions
A number of specialized resources have been developed for the reconstruction and analysis of plant gene networks. These include PlantRegMap (Tian et al., 2020), designed for analyzing transcription factor regulatory interactions; STRING (Szklarczyk et al., 2021), which enables the exploration of protein–protein interactions; the KEGG PLANT platform (Kanehisa, 2013), which integrates information on metabolic pathways across various plant species; and the Plant Reactome resource (Naithani et al., 2020), containing detailed data on signaling and metabolic pathways in model plant organisms. For visualization and analysis of gene networks, the Cytoscape software environment (Otasek et al., 2019) is widely used, offering an extensive set of plugins for working with biological data. The ncPlantDB database provides comprehensive information for analyzing regulatory networks, including data on cell-type specific expression of noncoding RNAs and their interactions (Cheng et al., 2024; Liu et al., 2025). The integration of such omics resources forms an effective platform for reconstructing gene networks of agricultural crops (Chao et al., 2023).
Earlier, we developed the ANDSystem cognitive software information platform (Ivanisenko V.A. et al., 2015, 2019; Ivanisenko T.V., 2020, 2022) designed for the full knowledge-engineering cycle in the biomedical domain. The system’s knowledge base contains more than 50 million interactions for various organisms
In the field of plant biology, ANDSystem has been used to create a knowledge base on the genetics of Solanum tuberosum (Saik et al., 2017; Ivanisenko T.V. et al., 2018; Demenkov et al., 2019), to reconstruct and analyze the regulatory gene network controlling cell wall functions in Arabidopsis thaliana leaves under water deficit (Volyanskaya et al., 2023), and to develop a method for prioritizing biological processes based on the reconstruction and analysis of associative gene networks (Demenkov et al., 2021).
The application of the ANDSystem automated reconstruction of associative gene networks to analyze microRNAmediated regulation of bread wheat (Triticum aestivum L.) adaptation to water deficit made it possible to propose new candidate microRNAs (MIR7757, MIR9653a, MIR9670, MIR9672b) of interest for further experimental studies of plant adaptation mechanisms under insufficient moisture (Kleshchev et al., 2024).
In another study (Antropova et al., 2024), ANDSystem was used to reconstruct the molecular genetic network of rice (Oryza sativa) responses to Rhizoctonia solani infection under nitrogen excess, which revealed three potential mechanisms explaining reduced plant resistance to the pathogen. Key regulatory pathways were identified: an OsGSK2-mediated cascade, the OsMYB44–OsWRKY6–OsPR1 signaling pathway, and a pathway involving SOG1, Rad51, and the PR1/PR2 genes. In addition, markers promising for breeding were identified: 7 genes regulating a broad range of stress responses and 11 genes that modulate the immune system. Additional analysis of noncoding RNAs (Antropova et al., 2024) identified 30 microRNAs targeting genes within the reconstructed gene network. For two of them (Osa-miR396 and Osa-miR7695), approximately 7,400 unique long noncoding RNAs with differing co-expression indices were found, which may indicate a complex architecture of posttranscriptional regulation under nitrogen stress.
The aim of the present work was to adapt ANDSystem to create the SmartCrop knowledge base, integrating data on molecular genetic mechanisms and associative gene networks of stress responses in rice and wheat based on intelligent analysis of scientific publications and curated factual databases. This work included the development of a domain ontology and the optimization of intelligent knowledge-extraction methods from scientific texts using semantic–linguistic patterns and pretrained large language models. The SmartCrop ontology is represented by a set of interconnected dictionaries describing: molecular genetic entities (genes, proteins, metabolites, microRNAs), biological processes, phenotypic traits and diseases, pathogens, genetic biomarkers, markers of resistance to crop protection products, molecular targets of chemical crop protection agents, biotic and abiotic factors, crop protection products, as well as cultivars with their economically valuable and consumer traits.
As a result of automated analysis of scientific publications, the SmartCrop knowledge base was formed, integrating more than 10 million interactions among the entities defined in the ontology
Materials and methods
Information resources used in the development of SmartCrop. To create the SmartCrop knowledge base, we used the ANDSystem software information platform (Ivanisenko V.A. et al., 2015, 2019; Ivanisenko T.V., 2020, 2022) and its information and bioinformatics technologies
Customization of ANDSystem methods for the subject domain was carried out using an ontology that included specialized dictionaries of entities and a description of the types of their interactions. The main sources of genetic and genomic information for constructing the dictionaries were: the NCBI Gene database (https://www.ncbi.nlm.nih. gov/gene), the rice-specific database Oryzabase (https:// shigen.nig.ac.jp/rice/oryzabase), the microRNA database miRBase (https://www.mirbase.org), the long noncoding RNA co-expression database ncPlantDB (https://bis.zju. edu.cn/ncPlantDB/ ), the single nucleotide polymorphism database dbSNP (https://www.ncbi.nlm.nih.gov/snp), and the database on cereal crops GrainGenes (https://wheat. pw.usda.gov/GG3)
To standardize terminology, we used the following ontologies: Gene Ontology (http://geneontology.org), Crop Ontology for wheat and rice (https://cropontology.org), as well as the genetic resources collection of VIR (https:// www.vir.nw.ru).
Chemical compounds and metabolites were annotated using the ChEBI database (https://www.ebi.ac.uk/chebi). Information on herbicide resistance was obtained from the International Herbicide-Resistant Weed Database (http://www.weedscience.org), and data on pesticides were taken from the EU Pesticide Database (https://food. ec.europa.eu/plants/pesticides/eu-pesticides-database_en). For knowledge extraction from texts, we used ANDSystem’s semantic-linguistic templates, as well as newly developed templates tailored to the specifics of the subject domain. In addition, artificial intelligence methods were applied for knowledge extraction, including GraphSAGE graph neural networks (Hamilton et al., 2017) and the large language model Gemma-2-9b-it (Gemma Team, Google DeepMind, 2024).
Evaluation of accuracy. To assess the quality of namedentity annotation in the text, the F1-score was used, which is the harmonic mean of precision (Precision) and recall (Recall):
F1 = 2·(Precision × Recall)/(Precision + Recall), Precision = TP/(TP + FP), Recall = TP/(TP + FN), where TP – are true positives, FP – are false positives, and FN – are false negatives.
Results
A schematic representation of the main components of the SmartCrop software-information system is shown in Figure 1.
Schematic representation of the architecture of the SmartCrop software information system.
SmartCrop domain-specific ontology module
The development of a domain-specific ontology was a key stage in building SmartCrop. The domain-oriented ontology defines a conceptual model of the problem area and includes dictionaries of entities and types of their interactions. Based on these dictionaries, information about interactions between specific entities is extracted from texts and factual databases. The current version of the SmartCrop ontology contains 15 dictionaries of different entity types (Table 1), compiled by extracting entity names from specialized databases and existing ontologies.
Dictionaries represented in the SmartCrop ontology
Interaction types. In the SmartCrop system, 16 types of relationships between ontology entities are defined. All interactions in the system are directional and can be divided into several main groups. Physical interactions include processes of forming both short-lived molecular complexes and stable associations between proteins and metabolites.
Chemical interactions comprise catalytic reactions of the substrate–enzyme–product type, protein proteolysis, as well as various post-translational protein modifications such as phosphorylation and glycosylation
A distinct group is formed by regulatory interactions, which encompass the regulation of gene expression by transcription factors, modulation of protein activity and function, control of protein and metabolite transport, as well as regulation of protein stability and degradation. An important feature is that regulatory interactions also define relationships between molecular genetic entities, biological processes, and phenotypic traits. Each regulatory event may be characterized by an enhancing or attenuating effect on the corresponding process.
Expression and co-expression of genes are distinguished separately. The products of gene expression are proteins and noncoding RNAs. Co-expression is the simultaneous expression of genes driven by shared regulatory mechanisms under changing cellular conditions. Additionally, the system accounts for associative links, which include unclassified interactions between various ontology entities.
Text-annotation module based on ontology entities
Recognition of molecular genetic entities in scientific texts is a challenging task due to the specific nature of their nomenclature. Our experience with ANDSystem shows that a substantial portion of errors in automatic reconstruction of associative gene networks is associated with inaccurate identification of named entities (Ivanisenko T.V. et al., 2022). The causes of such errors include the use of abbreviations by authors, semantic ambiguity of terms, and various linguistic features of scientific texts. In publications, standard names of entities are often modified, punctuation and word order are altered, grammatical forms vary, abbreviations are used, or technical typos are introduced (Pearson, 2001; Krallinger et al., 2015; Islamaj et al., 2021).
To improve recognition accuracy, we developed a twostage process: 1) initial matching of names to the ontology dictionary and 2) subsequent verification of whether each annotated entity name corresponds to its type, based on contextual document analysis using neural networks.
The verification process is implemented as follows: a language model converts the context (about 400 words) containing the analyzed entity, which is replaced with a special mask tag, into a vector representation. Based on this representation, a neural network performs binary classification, determining whether the contextual environment of the term is consistent with its typical usage
For entities from the ANDSystem ontology (genes, proteins, metabolites, etc.), classification accuracy was reported in a previously published paper (Ivanisenko T.V. et al., 2022). For the new SmartCrop dictionaries, manual expert evaluation of annotation quality was carried out (Table 2) based on the analysis of 1,000 randomly selected documents from the PubMed and PubMed Central databases
Accuracy assessment of entity name annotation for the new dictionaries
The evaluation results demonstrated high annotation accuracy for most dictionaries, with the exception of rice and wheat cultivar names. The identification of plant cultivar names is a well-known complex task, determined by several factors, including substantial overlap of terms with common vocabulary and anthroponyms, as well as the lack of a unified standard in the nomenclature of new cultivars (Do et al., 2018; D’Souza, 2024).
To address this problem, a specialized language model was trained, focused on the task of contextual term classification. The training was carried out in accordance with the methodology previously described in our work on improving the accuracy of identifying eight types of molecular genetic entities, including proteins, genes, metabolites, and cellular components (Ivanisenko T.V. et al., 2022). Integration of the developed model made it possible to substantially increase the recognition accuracy (F1-score) of cultivar names to 0.88 for rice and 0.919 for wheat.
Knowledge extraction module
The knowledge extraction module for scientific texts implements three main stages: 1) primary knowledge extraction using semantic-linguistic templates; 2) reconstruction of the initial semantic network; 3) its extension using graph neural networks and large language models. Additionally, to further expand the semantic network, a data extraction module for factual databases containing structured information is used, which makes it possible to obtain additional information about interactions between entities.
Semantic-linguistic templates are structured records containing metadata about the types of entities and the nature of their interactions. They include two main components:
- syntactic relations that describe the order of entities and keywords in a sentence using regular expressions, and
- semantic relations that define the type of interaction between entities. Regular expressions are used to search for patterns in the arrangement of entity names in annotated text sentences. When a match is found, specific entity names from the text are mapped to the template identifiers.
For each interaction type, specialized groups of templates with unique syntactic rules were developed. The knowledge base contains more than 18,000 ANDSystem templates for interaction types represented in both the ANDSystem and SmartCrop ontologies, as well as more than 3,000 templates specifically designed for the rice and wheat ontologies. The effectiveness of the template-based interaction extraction method was demonstrated during the development of ANDSystem (Ivanisenko V.A. et al., 2015).
Application of graph neural networks and large language models. At the second stage, based on the knowledge extracted using templates, a primary knowledge graph (semantic network) was constructed and used to train a graph neural network. After training, the network was used to predict missing edges in the knowledge graph. At the third stage, large language models were applied to validate these predictions by analyzing scientific texts in which the annotated entities with the predicted interactions co-occur (Ivanisenko T.V. et al., 2024).
Integration with omics data
Noncoding RNAs (ncRNAs) represent a broad and functionally diverse class of RNA molecules that are not translated into proteins but perform key regulatory functions in the cell. Long noncoding RNAs (lncRNAs) are of particular inte- rest, as they participate in the regulation of gene expression at multiple levels – from modulating mRNA stability and translation to being involved in complex signaling cascades (Statello et al., 2021; Supriya et al., 2024).
A well-known specialized resource on ncRNA co-expression in plants, including rice lncRNAs, is the ncPlantDB database (https://bis.zju.edu.cn/ncPlantDB/). It provides information on tissue-specific ncRNA expression at the single-cell level and their putative interactions, obtained using modern single-cell transcriptomics methods (Cheng et al., 2024; Liu et al., 2025). Integration of SmartCrop with ncPlantDB made it possible to use ncRNA co-expression data, including their relationships with microRNAs, to enrich the reconstructed gene networks.
Module for gene network analysis and visualization
As the graphical user interface of SmartCrop, intended for the reconstruction and analysis of gene networks based on information from the SmartCrop knowledge base, the ANDVisio software is used (Fig. 2).
Screenshot of the ANDVisio program interface.
The ANDVisio program (Demenkov et al., 2012) was originally developed as a component of the ANDSystem platform and was later adapted for integration with Smart- Crop. It provides researchers with a wide range of tools for structural and functional analysis of gene networks, including:
• multiple graph layout algorithms; • a multi-parameter filtering system; • mechanisms for pathways and cycles finding; • tools for calculating node centrality measures; • tools for assessing the enrichment of biological processes with network genes; • additional methods of network analysis
SmartCrop knowledge base
The system’s knowledge base is implemented as a semantic network (knowledge graph) that integrates data extracted both from scientific publications and from factual databases. In this graph structure, nodes correspond to entities of the domain ontology, and edges represent various types of interactions between them
The knowledge base was populated through systematic analysis of the scientific literature, including abstracts from PubMed and full-text articles from the open-access resource PubMed Central. The time span of the analyzed publications covered the period from 1970 to 2024, with the main selection criterion being the presence of references to wheat or rice. Detailed statistics on the number of recorded interactions in the SmartCrop knowledge base are presented in Table 3.
Statistics of the SmartCrop knowledge base on interactions between entities for wheat and rice
Discussion
To demonstrate the capabilities of SmartCrop, we consider two use cases: analysis of experimental omics data and experiment planning.
Analysis of experimental omics data
As an example of omics data interpretation, we performed functional annotation of differentially expressed genes (DEGs) in bread wheat under salt stress. For the analysis, we used a set of 5,829 DEGs obtained from the NCBI GEO database (GSE225565, Alyahya, Taybi, 2023) for root tissues of bread wheat (Triticum aestivum L., cultivar Saudi) in response to salinization
The results of the overrepresentation analysis of Smart- Crop entities (biological processes, phenotypic traits, agronomically important traits, pathogens) for this DEG set and their protein products are presented in Supplementary Table S11. In total, significant overrepresentation (p-value < 0.05, Bonferroni-corrected) was found for 217 terms describing biological processes (entity type Pathway), 50 phenotypic traits (Phenotype), 9 agronomically important traits (Agrophenotype), and 38 pathogenic species. The list of entities belonging to the five groups of the most statistically significant characteristics is given in Table 4.
Supplementary Materials are available in the online version of the paper: https://vavilovj-icg.ru/download/pict-2025-29/appx46.xls
Bread-wheat characteristics significantly associated with the DEG set and their protein products under salt stress, identified using the SmartCrop system
Analysis of the overrepresented biological processes showed that the DEG set under study is associated not only with the response to salt stress, but also with the response to water deficit. This reflects plant adaptation mechanisms to the state of so-called “physiological drought”, which arises when effective water uptake becomes impossible due to high osmotic pressure of the surrounding environment. Among such adaptations is stomatal closure, mediated by a rapid increase in abscisic acid levels (Verma et al., 2016; Zhao et al., 2021). Accordingly, the significantly overrepresented entities included both the phenotypic trait “stomatal closure rate” (Table 4) and signaling pathways associated with abscisic acid (Table S1), which confirms their important role in the response to salinity
It should be noted that an important advantage of the SmartCrop knowledge base, compared with widely used resources for gene functional annotation (DAVID, Gene Ontology, ShinyGO, etc.), is the ability to analyze relationships between genes/proteins and not only biological processes, molecular functions, cellular components, and KEGG pathways, but also a broad spectrum of abiotic and biotic environmental factors, phenotypic traits, agronomically important properties, and pathogens. This integration makes it possible to assess overrepresentation for different types of entities in the gene set under study, substantially expanding the capabilities of functional annotation and enabling the identification of genes with pleiotropic effects. The latter is particularly important for marker-assisted selection, since selection based on a single target phenotypic trait or genetic marker may simultaneously affect several other, non-target traits.
In particular, the results of functional annotation of DEGs in bread wheat under salt stress showed their association not only with responses to salinity and water deficit, but also with seed germination and with agronomically important traits reflecting grain quality (Table 4). For example, aquaporins (encoded by genes LOC543267, LOC100037645, LOC123093445, and others) provide selective transport of water molecules, participate in maintaining cellular ion balance and in regulating water–salt homeostasis under elevated salinity (Ayadi et al., 2019), and also facili- tate the movement of water and solutes within seeds, which plays a key role in the germination process (Hoai et al., 2020).
The functionality of SmartCrop is not limited to overrepresentation analysis. The system also makes it possible to reconstruct associative networks of proteins and genes significantly associated with overrepresented entities and to search for their regulators. This provides a deeper understanding of the molecular mechanisms underlying these relationships and helps to reveal their specificity under experimental conditions.
As an example, a gene network regulating plant tolerance to hyperosmotic stress (GO:0042538 hyperosmotic salinity response) was reconstructed (Fig. 3). According to SmartCrop, the wheat response to hyperosmotic stress involves 95 genes and 119 proteins, including aquaporins and sodium/hydrogen exchangers, which play a key role in regulating intracellular pH, water balance, and sodium-ion homeostasis (Gupta et al., 2021). Excess sodium ions entering from the environment are removed from the cytoplasm into the apoplast and vacuoles in exchange for hydrogen ions via transmembrane Na+/H+ exchangers (Zhao et al., 2021).
Genes, proteins, and microRNAs involved in regulating the response of bread wheat to hyperosmotic stressGenes differentially expressed in bread-wheat roots in response to soil salinization, as well as their protein products, are highlighted with a green frame
The network also includes peroxidases and catalases involved in antioxidant defense under abiotic stress; transcription factors of the MYB and WRKY families; dehydrins (LOC123125487, LOC100141381, and others); cold-shock proteins (LOC123080042, LOC543252, LOC542792); as well as DELLA proteins, which, by suppressing the gibberellin signaling pathway and interacting with jasmonic acid signaling, increase plant tolerance to abiotic stress, including salinity (Colebrook et al., 2014). In addition, the network contains calcium-dependent protein kinases – key components of calcium signaling cascades activated under abiotic stress
In addition, according to SmartCrop, the regulation of the response to hyperosmotic stress involves the microRNA tae- MIR159a, which regulates the expression of the transcription factor TaMyb3 (LOC543161), as well as tae-MIR1122b and tae-MIR9668, the targets of which are the aquaporins LOC123054192 and LOC123093495, respectively.
Of the full set of genes involved in the regulation of the hyperosmotic stress response, only nine showed differential expression in bread-wheat root tissues under experimental salt stress in the study (Alyahya, Taybi, 2023). This list includes genes encoding aquaporins, peroxidases, catalases, and the serine/threonine protein kinase CTR1 (LOC100286402). Thus, under the experimental conditions described by (Alyahya, Taybi, 2023), signaling pathways associated primarily with antioxidant defense were activated
The associative network reconstructed in SmartCrop includes these nine DEGs and their protein products, regulatory proteins, as well as two microRNAs: tae- MIR159a, which regulates expression of the transcription factor TaMyb3 (LOC543161), and tae-MIR9668, targeting the aquaporin LOC123093495. This network is shown in Figure 4.
Gene network regulating the response of bread wheat to salt stress.Genes differentially expressed in bread-wheat roots in response to soil salinization, as well as their protein products, are highlighted with a green frame.
It is interesting to note that the transcription factor TaMyb3 (LOC543161), which is a target of the microRNA tae-MIR159a, in turn acts as a negative regulator of the expression of several genes encoding peroxidases. Suppression of the expression of these enzymes leads to increased accumulation of hydrogen peroxide in tissues and, consequently, to reduced plant tolerance to salinity (Wei et al., 2021). Thus, in this case a “cassette-cascade” regulatory principle involving microRNAs is implemented, in which a microRNA controls the expression of its target transcription factor, and the latter regulates an entire set of genes involved in the response to abiotic stress (Kleshchev et al., 2024).
Transcription factors of the MYB family are well known as regulators of responses to various abiotic stresses, including salinity (Kong et al., 2021; Wang S. et al., 2021). In particular, they participate in the regulation of flavonoid biosynthesis – metabolites required for protecting cells from oxidative stress (Wang X. et al., 2021).
Application of SmartCrop to experimental design
search for promising genes and phenotypic markers for subsequent marker-assisted selection and genome editing aimed at increasing rice (Oryza sativa L.) tolerance to soil salinity.
According to SmartCrop, the following traits can serve as markers of salinity tolerance: chlorophyll content, seed shape, and the content of the metabolites 3′-methoxyapigenin and 5,7,4′-trihydroxy-3′-methoxyflavone. According to the SmartCrop knowledge base, rice tolerance to salinity is regulated by 30 genes and their corresponding 30 protein products (Fig. 5). In addition to genes, this regulation involves the microRNAs osa-MIR444f and osa-MIR444e, which target the transcription factor OsMADS23, as well as osa-MIR444e, targeting the auxin receptor OsABF4. The transcription factor OsBBX11, a known regulator of salinity tolerance (Lei et al., 2023), is targeted by the microRNAs osa-MIR319a and osa-MIR396c.
Long noncoding RNAs (lncRNAs) are molecules longer than 200 nucleotides that regulate gene expression at the transcriptional, post-transcriptional, and epigenetic levels, thereby modulating plant responses to various abiotic and biotic factors, including salinity (Sun X. et al., 2018). lncRNAs can interact with DNA (chromatin, promoters, and enhancers), proteins, mRNAs, and microRNAs. One important mechanism of their action is binding to microRNAs, which prevents the latter from acting on their targets and thus turns lncRNAs into key regulators of microRNA activity (Saha et al., 2025).
According to SmartCrop, the microRNA osa-MIR396c interacts with 508 long noncoding RNAs, six of which (LNC-Os02g06395, LNC-Os03g08620, LNC-Os03g25810, LNC-Os07g13605, LNC-Os08g32435, LNC-Os09g33385) are co-expressed not only with osa-MIR396c but also with 42 other rice microRNAs (Fig. 5). This indicates their potential role as key players in the regulation of rice tolerance to abiotic stresses, including salinity.
Associative gene network illustrating the involvement of genes, proteins, microRNAs, and long noncoding RNAs in the regulation of rice (Oryza sativa L.) tolerance to salinity and their potential role as phenotypic markersGenes in rice and the proteins they encode that positively regulate both salt tolerance and other agronomically important traits are outlined in green. Genes in rice and the proteins they encode that enhance salt tolerance but suppress other agronomically important traits are outlined in red.
Of the 30 genes that regulate salinity tolerance, six (OsPIL13, OsNBL1, OsABF4, OsCPK10, OsCRT3, OsBBX11) control chlorophyll content. The remaining 24 genes have not previously been associated with known markers of rice salinity tolerance and therefore represent promising candidates for the discovery of new genetic markers of this trait
It should be particularly emphasized that prioritizing genes for marker-assisted selection and genome editing requires consideration of the specificity of their regulatory effects, since selection for a single target trait may influence other agronomically important characteristics. The analysis showed that genes and proteins regulating salinity tolerance are associated with 67 other phenotypic traits, including biomass, leaf area, grain morphology, and others, which reflects pleiotropic effects.
Three genes – OsPIL13, Ehd1, and OsGA2Ox3 – are positive regulators of both salinity tolerance and such agronomically important traits as grain quality, seed dormancy period, and grain length. This makes them promising candidates for breeding and genome editing, since their modulation may simultaneously increase salt tolerance and improve grain quality. At the same time, the genes OsWRKY63, OsRAM2, and OsABF4 enhance rice tolerance to salinity but are associated with negative regulation of seed dormancy period, grain protein content, and plant resistance to Fusarium graminearum and F. pseudograminearum, which must be considered in breeding programs.
According to SmartCrop, 21 genes are involved exclusively in the regulation of salinity tolerance and are not associated with the regulation of agronomically important traits or resistance to pathogens, which makes them suitable candidates for targeted breeding aimed at increasing salt tolerance.
Another important factor that must be taken into account when selecting genes for marker-assisted selection and/ or genome editing is the potential bidirectionality of their regulatory effects, since gene products may either stimulate or suppress biological processes involved in the positive or negative regulation of the target trait. To assess such bidirectionality, the “Pathway Wizard” module of the ANDSystem program was used to identify regulatory relationships between the protein products of the 30 genes associated with rice salinity tolerance and the biological processes that, in turn, participate in regulating this trait (Fig. 6).
Regulatory relationships between genes associated with rice salinity tolerance and biological pathways involved in the regulation of this trait.
Among the genes regulating salinity tolerance, particular interest is drawn to OsMYB48-1, OsCPK10, OsCBL10, OsDREB2B, OsRAM2, and NBS-LRR, which exhibit a unidirectional effect in the form of positive regulation of key processes that ensure tolerance to salt stress (hyperosmotic salinity response, stomatal closure, ABA-independent pathway, etc.). The high degree of connectivity of these genes with the target trait, combined with the unidirectional nature of their regulatory action, suggests that their use in markerassisted selection or genome editing may have a more direct and pronounced impact on increasing salt tolerance compared with other candidates.
Conclusion
The SmartCrop knowledge base is a specialized version of the ANDSystem software information platform, adapted for the tasks of rice and wheat genetics and breeding. It integrates information on a wide range of entities – genes, proteins, metabolites, noncoding RNAs, biological processes, breeding-relevant and phenotypic traits, pathogens, as well as biotic and abiotic factors – and their relationships. This architecture provides extensive opportunities for studying the molecular genetic mechanisms of plant stress tolerance, as well as for selecting genes, genetic markers, and phenotypic traits within the framework of marker-assisted selection of crop plants
Examples of SmartCrop applications for the functional annotation of differentially expressed genes in bread wheat under salt stress and for planning experiments to increase rice salinity tolerance using marker-assisted selection have demonstrated the high efficiency of the system and its potential for solving applied problems in breeding and genome editing.
Conflict of interest
The authors declare no conflict of interest.
References
Alyahya N., Taybi T. Comparative transcriptomic profiling reveals differentially expressed genes and important related metabolic pathways in shoots and roots of a Saudi wheat cultivar (Najran) under salinity stress. Front Plant Sci. 2023;14:1225541. doi 10.3389/ fpls.2023.1225541
Antropova E.A., Volyanskaya A.R., Adamovskaya A.V., Demenkov P.S., Yatsyk I.V., Ivanisenko T.V., Orlov Y.L., Haoyu Ch., Chen M., Ivanisenko V.A. Computational identification of promising genetic markers associated with molecular mechanisms of reduced rice resistance to Rhizoctonia solani under excess nitrogen fertilization using gene network reconstruction and analysis methods. Vavilovskii Zhurnal Genetiki i Selektsii = Vavilov Journal of Genetics and Breeding. 2024;28(8):960-973. doi 10.18699/vjgb-24-103
Ayadi M., Brini F., Masmoudi K. Overexpression of a wheat aquaporin gene, TdPIP2;1, enhances salt and drought tolerance in transgenic durum wheat cv. Maali. Int J Mol Sci. 2019;20(10):2389. doi 10.3390/ijms20102389
Chao H., Zhang S., Hu Y., Ni Q., Xin S., Zhao L., Ivanisenko V.A., Orlov Y.L., Chen M. Integrating omics databases for enhanced crop breeding. J Integr Bioinform. 2023;20(4):20230012. doi 10.1515/ jib-2023-0012
Chen T., Nomura K., Wang X., Sohrabi R., Xu J., Yao L., Paasch B.C., Ma L., Kremer J., Cheng Y., Zhang L., Wang N., Wang E., Xin X.F., He S.Y. A plant genetic network for preventing dysbiosis in the phyllosphere. Nature. 2020;580(7805):653-657. doi 10.1038/s41586- 020-2185-0
Cheng M., Zhu Y., Yu H., Shao L., Zhang Y., Li L., Tu H., … Orlov Y.L., Chen D., Wong A., Yang Y.E., Chen M. Non-coding RNA notations, regulations and interactive resources. Funct Integr Genomics. 2024;24(6):217. doi 10.1007/s10142-024-01494-w
Colebrook E.H., Thomas S.G., Phillips A.L., Hedden P. The role of gibberellin signalling in plant responses to abiotic stress. J Exp Biol. 2014;217(1):67-75. doi 10.1242/jeb.089938
Demenkov P.S., Ivanisenko T.V., Kolchanov N.A., Ivanisenko V.A. ANDVisio: a new tool for graphic visualization and analysis of literature mined associative gene networks in the ANDSystem. In Silico Biol. 2012;11(3-4):149-161. doi 10.3233/isb-2012- 0449
Demenkov P.S., Saik O.V., Ivanisenko T.V., Kolchanov N.A., Kochetov A.V., Ivanisenko V.A. Prioritization of potato genes involved in the formation of agronomically valuable traits using the SOLANUM TUBEROSUM knowledge base. Vavilovskii Zhurnal Genetiki i Selektsii = Vavilov Journal of Genetics and Breeding. 2019; 23(3):312-319. doi 10.18699/VJ19.501
Demenkov P.S., Oshchepkova E.A., Ivanisenko T.V., Ivanisenko V.A. Prioritization of biological processes based on the reconstruction and analysis of associative gene networks describing the response of plants to adverse environmental factors. Vavilovskii Zhurnal Genetiki i Selektsii = Vavilov Journal of Genetics and Breeding. 2021;25(5):580-592. doi 10.18699/VJ21.065
Do H., Than K., Larmande P. Evaluating named-entity recognition approaches in plant molecular biology. In: Multi-disciplinary Trends in Artificial Intelligence. MIWAI 2018. Lecture Notes in Computer Science. Vol. 11248. Springer, Cham., 2018;219-225. doi 10.1007/ 978-3-030-03014-8_19
D’Souza J. Agriculture named entity recognition – towards FAIR, reusable scholarly contributions in agriculture. Knowledge. 2024; 4(1):1-26. doi 10.3390/knowledge4010001
Gemma Team, Google DeepMind. Gemma 2: improving open language models at a practical size. arXiv. 2024:2408.00118. doi 10.48550/ arXiv.2408.00118
Guindani L.G., Oliveirai G.A., Ribeiro M.H.D.M., Gonzalez G.V., de Lima J.D. Exploring current trends in agricultural commodities forecasting methods through text mining: developments in statistical and artificial intelligence methods. Heliyon. 2024;10(23):e40568. doi 10.1016/j.heliyon.2024.e40568
Gupta A., Shaw B.P., Sahu B.B. Post-translational regulation of the membrane transporters contributing to salt tolerance in plants. Funct Plant Biol. 2021;48(12):1199-1212. doi 10.1071/FP21153
Hamilton W.L., Ying R., Leskovec J. Inductive representation learning on large graphs. arXiv. 2017. doi 10.48550/arxiv.1706.02216
Hoai P.T.T., Tyerman S.D., Schnell N., Tucker M., McGaughey S.A., Qiu J., Groszmann M., Byrt C.S. Deciphering aquaporin regulation and roles in seed biology. J Exp Bot. 2020;71(6):1763-1773. doi 10.1093/jxb/erz555
Islamaj R., Wei C.H., Cissel D., Miliaras N., Printseva O., Rodionov O., Sekiya K., Ward J., Lu Z. NLM-Gene, a richly annotated gold standard dataset for gene entities that addresses ambiguity and multi-species gene recognition. J Biomed Inform. 2021;118:103779. doi 10.1016/j.jbi.2021.103779
Ivanisenko T.V., Demenkov P.S., Ivanisenko V.A. Text mining on PubMed. In: Chen M., Hofestädt R. (Eds) Approaches in Integrative Bioinformatics. Springer, 2014;161-170. doi 10.1007/978-3-642- 41281-3_6
Ivanisenko T.V., Saik O.V., Demenkov P.S., Khlestkin V.K., Khlestkina E.K., Kolchanov N.A., Ivanisenko V.A. The SOLANUM TUBEROSUM knowledge base: the section on molecular-genetic regulation of metabolic pathways. Vavilovskii Zhurnal Genetiki i Selektsii = Vavilov Journal of Genetics and Breeding. 2018;22(1): 8-17. doi 10.18699/VJ18.325 (in Russian)
Ivanisenko T.V., Saik O.V., Demenkov P.S., Ivanisenko N.V., Savostianov A.N., Ivanisenko V.A. ANDDigest: a new web-based module of ANDSystem for the search of knowledge in the scientific literature. BMC Bioinformatics. 2020;21(S11):228. doi 10.1186/s12859-020- 03557-8
Ivanisenko T.V., Demenkov P.S., Kolchanov N.A., Ivanisenko V.A. The new version of the ANDDigest tool with improved AI-based short names recognition. Int J Mol Sci. 2022;23(23):14934. doi 10.3390/ ijms232314934
Ivanisenko T.V., Demenkov P.S., Ivanisenko V.A. An accurate and efficient approach to knowledge extraction from scientific publications using structured ontology models, graph neural networks, and large language models. Int J Mol Sci. 2024;25(21):11811. doi 10.3390/ ijms252111811
Ivanisenko V.A., Saik O.V., Ivanisenko N.V., Tiys E.S., Ivanisenko T.V., Demenkov P.S., Kolchanov N.A. ANDSystem: an Associative Network Discovery System for automated literature mining in the field of biology. BMC Syst Biol. 2015;9(Suppl.2):S2. doi 10.1186/1752- 0509-9-s2-s2
Ivanisenko V.A., Demenkov P.S., Ivanisenko T.V., Mishchenko E.L., Saik O.V. A new version of the ANDSystem tool for automatic extraction of knowledge from scientific publications with expanded functionality for reconstruction of associative gene networks by considering tissue-specific gene expression. BMC Bioinformatics. 2019;20(S1):34. doi 10.1186/s12859-018-2567-6
Kanehisa M. Molecular network analysis of diseases and drugs in KEGG. In: Mamitsuka H., DeLisi C., Kanehisa M. (Eds) Data Mining for Systems Biology. Methods in Molecular Biology. Vol. 939. Humana Press, 2013;263-275. doi 10.1007/978-1-62703- 107-3_17
Kleshchev M.A., Maltseva A.V., Antropova E.A., Demenkov P.S., Ivanisenko T.V., Orlov Y.L., Chao H., Chen M., Kolchanov N.A., Ivanisenko V.A. Reconstruction and computational analysis of the microRNA regulation gene network in wheat drought response mechanisms. Vavilovskii Zhurnal Genetiki i Selektsii = Vavilov Journal of Genetics and Breeding. 2024;28(8):904-917. doi 10.18699/ vjgb-24-98
Kong W., Sun T., Zhang C., Deng X., Li Y. Comparative transcriptome analysis reveals the mechanisms underlying differences in salt tolerance between indica and japonica rice at seedling stage. Front Plant Sci. 2021;12:725436. doi 10.3389/ fpls.2021.725436
Krallinger M., Rabal O., Leitner F., Vazquez M., Salgado D., Lu Z., Leaman R., … Alves R., Segura-Bedmar I., Martínez P., Oyarzabal J., Valencia A. The CHEMDNER corpus of chemicals and drugs and its annotation principles. J Cheminform. 2015;7(S1):S2. doi 10.1186/1758-2946-7-s1-s2
Lei L., Cao L., Ding G., Zhou J., Luo Y., Bai L., Xia T., … Xie T., Yang G., Wang X., Sun S., Lai Y. OsBBX11 on qSTS4 links to salt tolerance at the seeding stage in Oryza sativa L. ssp. Japonica. Front Plant Sci. 2023;14:1139961. doi 10.3389/fpls.2023.1139961
Lesk C., Rowhani P., Ramankutty N. Influence of extreme weather disasters on global crop production. Nature. 2016;529(7584):84-87. doi 10.1038/nature16467
Liu L., Liu E., Hu Y., Li S., Zhang S., Chao H., Hu Y., Zhu Y., Chen Y., Xie L., Shen Y., Wu L., Chen M. ncPlantDB: a plant ncRNA database with potential ncPEP information and cell type-specific interaction. Nucleic Acids Res. 2025;53(D1):D1587-D1594. doi 10.1093/ nar/gkae1017
Mittler R. Abiotic stress, the field environment and stress combination. Trends Plant Sci. 2006;11(1):15-19. doi 10.1016/j.tplants. 2005.11.002
Naithani S., Gupta P., Preece J., D’Eustachio P., Elser J.L., Garg P., Dikeman D.A., … Bolton E., Papatheodorou I., Stein L., Ware D., Jaiswal P. Plant Reactome: a knowledgebase and resource for comparative pathway analysis. Nucleic Acids Res. 2020;48(D1):D1093- D1103. doi 10.1093/nar/gkz996
Ni Y., Aghamirzaie D., Elmarakeby H., Collakova E., Li S., Grene R., Heath L.S. A machine learning approach to predict gene regulatory networks in seed development in Arabidopsis. Front Plant Sci. 2016;7:1936. doi 10.3389/fpls.2016.01936
Nykiel M., Gietler M., Fidler J., Prabucka B., Labudda M. Abiotic stress signaling and responses in plants. Plants. 2023;12(19):3405. doi 10.3390/plants12193405
Otasek D., Morris J.H., Bouças J., Pico A.R., Demchak B. Cytoscape Automation: empowering workflow-based network analysis. Genome Biol. 2019;20(1):185. doi 10.1186/s13059-019-1758-4
Pearson H. Biology’s name game. Nature. 2001;411(6838):631-632. doi 10.1038/35079694
Saha C., Saha S., Bhattacharyya N.P. LncRNAOmics: a comprehensive review of long non-coding RNAs in plants. Genes. 2025;16(7):765. doi 10.3390/genes16070765
Saik O.V., Demenkov P.S., Ivanisenko T.V., Kolchanov N.A., Ivanisenko V.A. Development of methods for automatic extraction of knowledge from texts of scientific publications for the creation of a knowledge base SOLANUM TUBEROSUM. Agric Biol. 2017;52(1):63-74. doi 10.15389/agrobiology.2017.1.63eng
Shewry P.R., Hey S.J. The contribution of wheat to human diet and health. Food Energy Secur. 2015;4(3):178-202. doi 10.1002/fes3.64
Shrestha A.M.S., Gonzales M.E.M., Ong P.C.L., Larmande P., Lee H.S., Jeung J.U., Kohli A., Chebotarov D., Mauleon R.P., Lee J.S., McNally K.L. RicePilaf: a post-GWAS/QTL dashboard to integrate pangenomic, coexpression, regulatory, epigenomic, ontology, pathway, and text-mining information to provide functional insights into rice QTLs and GWAS loci. GigaScience. 2024;13:giae013. doi 10.1093/gigascience/giae013
Statello L., Guo C.J., Chen L.L., Huarte M. Gene regulation by long non-coding RNAs and its biological functions. Nat Rev Mol Cell Biol. 2021;22:96-118. doi 10.1038/s41580-020-00315-9
Sun X., Zheng H., Sui N. Regulation mechanism of long non-coding RNA in plant response to stress. Biochem Biophys Res Commun. 2018;503(2):402-407. doi 10.1016/j.bbrc.2018.07.072
Supriya P., Srividya G.K., Solanki M., Manvitha D., Prakasam V., Balakrishnan M., Neeraja C.N., Rao C.S., Sundaram R.M., Mangrauthia S.K. Identification and expression analysis of long noncoding RNAs of rice induced during interaction with Rhizoctonia solani. Physiol Mol Plant Pathol. 2024;134:102389. doi 10.1016/ j.pmpp.2024.102389
Szklarczyk D., Gable A.L., Nastou K.C., Lyon D., Kirsch R., Pyysalo S., Doncheva N.T., Legeay M., Fang T., Bork P., Jensen L.J., von Mering C. The STRING database in 2021: customizable protein– protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021;49(D1):D605- D612. doi 10.1093/nar/gkaa1074
Tian F., Yang D.C., Meng Y.Q., Jin J., Gao G. PlantRegMap: charting functional regulatory maps in plants. Nucleic Acids Res. 2020; 48(D1):D1104-D1113. doi 10.1093/nar/gkz1020Vaswani A., Shazeer N., Parmar N., Uszkoreit J., Jones L., Gomez A.N., Kaiser L., Polosukhin I. Attention is all you need. arXiv. 2017. doi 10.48550/arXiv.1706.03762
Verma V., Ravindran P., Kumar P.P. Plant hormone-mediated regulation of stress responses. BMC Plant Biol. 2016;16(1):86. doi 10.1186/ s12870-016-0771-y
Virlouvet L., Avenson T.J., Du Q., Zhang C., Liu N., Fromm M., Avramova Z., Russo S.E. Dehydration stress memory: gene networks linked to physiological responses during repeated stresses of Zea mays. Front Plant Sci. 2018;9:1058. doi 10.3389/fpls.2018. 01058
Volyanskaya A.R., Antropova E.A., Zubairova U.S., Demenkov P.S., Venzel A.S., Orlov Y.L., Makarova A.A., Ivanisenko T.V., Gorshkova T.A., Aglyamova A.R., Kolchanov N.A., Chen M., Ivanisenko V.A. Reconstruction and analysis of the gene regulatory network for cell wall function in Arabidopsis thaliana L. leaves in response to water deficit. Vavilov J Genet Breed. 2023;27(8):1031-1041. doi 10.18699/vjgb-23-118
Wang S., Shi M., Zhang Y., Xie X., Sun P., Fang C., Zhao J. FvMYB24, a strawberry R2R3-MYB transcription factor, improved salt stress tolerance in transgenic Arabidopsis. Biochem Biophysical Res Commun. 2021;569:93-99. doi 10.1016/j.bbrc.2021.06.085
Wang X., Niu Y., Zheng Y. Multiple functions of MYB transcription factors in abiotic stress responses. Int J Mol Sci. 2021;22(11):6125. doi 10.3390/ijms22116125
Wei T., Guo D., Liu J. PtrMYB3, a R2R3-MYB transcription factor from Poncirus trifoliata, negatively regulates salt tolerance and hydrogen peroxide scavenging. Antioxidants. 2021;10(9):1388. doi 10.3390/antiox10091388
Yan J., Wang X. Unsupervised and semi‐supervised learning: the next frontier in machine learning for plant systems biology. Plant J. 2022;111(6):1527-1538. doi 10.1111/tpj.15905
Zhang D., Zhao R., Xian G., Kou Y., Ma W. A new model construction based on the knowledge graph for mining elite polyphenotype genes in crops. Front Plant Sci. 2024;15:1361716. doi 10.3389/ fpls.2024.1361716
Zhao S., Zhang Q., Liu M., Zhou H., Ma C., Wang P. Regulation of plant responses to salt stress. Int J Mol Sci. 2021;22(9):4609. doi 10.3390/ijms22094609