Early-onset kidney failure in a girl with autosomal dominant tubulointerstitial kidney disease due to a de novo UMOD variant
Shinya Tomori, Kenichiro Miura, Yoko Shirai, Taeko Hashimoto, Ichiro Hada, Ryota Kurayama, Naoya Morisada, Kandai Nozu, Motoshi Hattori

TL;DR
A 7-year-old girl with no family history of kidney disease developed early-onset kidney failure due to a new mutation in the UMOD gene, highlighting the need to consider this condition in children.
Contribution
Identifies a de novo UMOD variant in a child with early-onset ADTKD, expanding understanding of its clinical presentation and genetic basis.
Findings
A de novo pathogenic variant c.172G > T in the UMOD gene was identified in a child with early-onset kidney failure.
The variant was located in the EGF-like domain 1, which may contribute to rapid progression to kidney failure.
ADTKD-UMOD should be considered in the differential diagnosis of childhood kidney failure, even without family history.
Abstract
Autosomal dominant tubulointerstitial kidney disease (ADTKD) is characterized by renal tubular and interstitial abnormalities and slow progressive loss of kidney function. Patients with ADTKD rarely progress to kidney failure in early childhood. A 7-year-old Japanese girl was admitted to the hospital due to afebrile seizures and was later diagnosed with Panayiotopoulos syndrome. Blood examinations showed that she had a serum creatinine level of 1.57 mg/dL (Cr-eGFR 28 mL/min/1.73 m²), consistent with chronic kidney disease stage 4. Ultrasonography showed bilateral small to normal-sized kidneys, with increased renal parenchymal echogenicity, poor corticomedullary differentiation, and small cysts. A panel exome sequencing targeting 187 genes identified a de novo pathogenic variant c.172G > T, p.Gly58Cys in the EGF-like domain 1 of the UMOD gene. Her parents did not possess this variant,…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
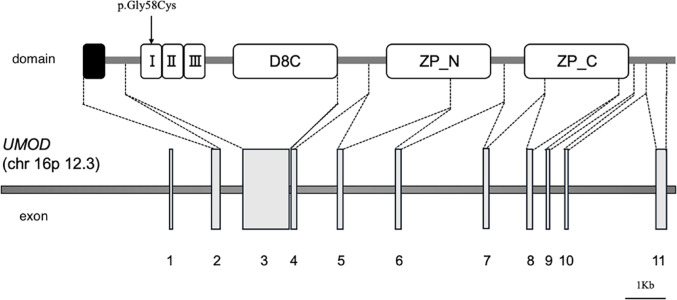 Figure 1
Figure 1- —Tokyo Women's Medical University
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsKidney Stones and Urolithiasis Treatments · Biomedical Research and Pathophysiology · Biochemical and Molecular Research
Introduction
Autosomal dominant tubulointerstitial kidney disease due to UMOD variants (ADTKD-UMOD) is a hereditary nephropathy caused by pathogenic variants in the UMOD gene, which encodes the glycoprotein uromodulin and is located on chromosome 16p12. Clinically, it is characterized by chronic kidney disease (CKD) and hyperuricemia [1]. CKD in ADTKD-UMOD typically progresses during adulthood, with a reported median age at diagnosis of 30.5 years [2] and a median age at onset of kidney failure of 47 years [3].While there have been reports of ADTKD identified during childhood through family screening at early stages of CKD, progression to kidney failure during childhood is rare [1]. Here, we report a sporadic pediatric case of ADTKD-UMOD diagnosed during the evaluation of advanced CKD.
Case Presentation.
A 7-year-old girl, born to healthy, non-consanguineous Japanese parents, was brought to the emergency department for afebrile seizure. The seizure resolved spontaneously, and brain MRI and EEG revealed no abnormalities. Subsequently, the seizures were diagnosed as Panayiotopoulos syndrome. As an incidental finding, her serum creatinine level was elevated at 1.57 mg/dL (creatinine-based eGFR: 28 mL/min/1.73 m²), consistent with CKD stage 4. She was referred to our hospital for further evaluation.
She had no history of abnormal urinalysis findings, and no family history of kidney disease or dialysis. Her height was 115 cm (–0.6 SD) and weight was 18.5 kg (–1.0 SD). No abnormalities were found on physical examination. Laboratory tests showed a creatinine-based eGFR of 26 mL/min/1.73 m², cystatin C-based eGFR of 31 mL/min/1.73 m², hemoglobin 9.3 g/dL, and uric acid 9.3 mg/dL. Early morning urine showed a specific gravity of 1.006, indicating hyposthenuria.
Ultrasonography indicated that the left kidney measured 6.3 × 3.9 cm (–2.3 SD) and the right kidney measured 6.9 × 3.9 cm (–1.6 SD), suggesting that the kidneys were small to normal in size. Increased echogenicity and the presence of several small cysts were also observed. These findings were suggestive of nephronophthisis. In addition, punctate echogenic foci consistent with calcifications were observed within the parenchyma. No abnormalities were found in the bladder, ureters, liver, or other organs. Otolaryngologic and ophthalmologic evaluations revealed no extrarenal manifestations.
Due to the sporadic nature of the case and the presence of normal to small-sized kidneys with cysts and progressive CKD in childhood, nephronophthisis was initially suspected. However, genetic testing for nephronophthisis-related genes, conducted with parental consent, revealed no pathogenic variants. Subsequently, targeted panel exome analysis covering 187 genes related to congenital anomalies of the kidney and urinary tract (CAKUT) and cystic kidney diseases was performed in the proband. Sanger sequencing was then conducted for the patient and both parents, and the variant was detected only in the patient but not in the parents. This confirmed that it was a de novo variant, c.172G > T, p.Gly58Cys, in the EGF-like domain 1 of the UMOD gene (Fig. 1) [4]. Neither parent carried this variant. Based on the results of genetic analysis, the patient was diagnosed with ADTKD-UMOD. Treatment for CKD was initiated with oral sodium bicarbonate and febuxostat. Subsequent monitoring of renal function revealed a decline in eGFR to 19 mL/min/1.73 m² 1 year and 2 months after the initial visit to our hospital, at which point the patient was registered for deceased donor kidney transplantation.
Fig. 1 Schematic representation of the UMOD gene structure and corresponding protein domains The UMOD protein, which consists of three epidermal growth factor (EGF)–like domains, a D8C domain, and a zona pellucida (ZP) domain. The variant identified in this case, p.Gly58Cys (arrow), is located within EGF-like domain 1, which is encoded by exon 3 I, EGF-like domain 1; II, EGF-like domain 2; III, EGF-like domain 3; D8C, central domain; ZP_N, N-terminal subdomain of the zona pellucida domain; ZP_C, C-terminal subdomain of the zona pellucida domain
Discussion
We present a rare pediatric sporadic case of ADTKD-UMOD with a de novo pathogenic variant in the EGF-like domain 1, associated with advanced CKD at an earlier age than previously reported. ADTKD-UMOD is typically characterized by CKD progression during adulthood. Bleyer et al. reported that among 34 patients with ADTKD-UMOD under the age of 18, none had renal dysfunction corresponding to CKD stage 4 or higher [1]. However, there have been reports of pediatric patients progressing to kidney failure [1, 4, 5]. This case adds to the literature suggesting that although rare, ADTKD-UMOD may present with kidney failure during childhood.
ADTKD is typically characterized by a positive family history, which is also reflected in the diagnostic criteria [6]. However, approximately 10% of ADTKD-UMOD cases occur without a family history [7]. Our case lacked a family history, and initial differential diagnoses included CAKUT and nephronophthisis. Genetic testing ultimately confirmed a de novo pathogenic UMOD variant. Therefore, even in the absence of a family history, ADTKD-UMOD should be considered in the differential diagnosis of pediatric advanced CKD.
Uromodulin consists of EGF-like domains, a central D8C domain, a zona pellucida domain, and a GPI anchor [8]. Although the mechanism by which UMOD variantscause kidney disease remains unclear, EGF-like domains are common protein motifs involved in cell adhesion, coagulation, and receptor-ligand interactions. Moskowitz et al. reported that patients with variants in EGF-like domains 2 or 3 progressed to kidney failure at a younger age than those with variants in other domains [9]. Zaucke et al. reported two patients with the same variant as in our case, both progressing to severe CKD by age 12 and 13, respectively (eGFR 3 and 23 mL/min/1.73 m²) [4]. To date, five cases of pediatric-onset kidney failure due to ADTKD-UMOD have been reported (Table 1) [1, 4, 5], with a median age of kidney failure onset at 12 years. Of these, three had variants in EGF-like domain 1. These findings suggest that variants in the EGF-like domain may be associated with early-onset kidney failure.
Table 1. Previously reported cases of ADTKD-UMOD with kidney failure during childhoodNo.Author, YearAge at Kidney FailureSexFamily HistoryNucleotide variantThe change of amino acid sequenceDomain1–1Zaucke, 2010 [4]13N/APresent (details unknown)c.172G > Tp.Gly58CysEGF11–212N/APresent (details unknown)c.172G > Tp.Gly58CysEGF12Carucci, 2019 [5]6MaleMother had the same variant (kidney failure at 31 years)c.249 C > Gp.Cys83TrpEGF23 − 1Bleyer, 2022 [1]12N/ARelatives had kidney failure at ages 18 and 24 (details unknown)c.155G > Cp.Cys52SerEGF13 − 214N/Ac.155G > Cp.Cys52SerEGF1The median age of kidney failure among these five cases was 12 years, and four out of the five patients had variants in the EGF-like domain 1
Conclusion
We report a sporadic pediatric case of ADTKD-UMOD identified through evaluation of advanced CKD. Although kidney failure during childhood is uncommon in ADTKD-UMOD, our case and previous reports indicate that variants in specific domains, such as the EGF-like domain, may lead to early disease progression. ADTKD-UMOD should be considered in the differential diagnosis of early-onset CKD and kidney failure in pediatric patients.
The reference list from the paper itself. Each links out to its DOI / PubMed record.