How much can reticulate evolution entangle plant systematics? Revisiting subfamilial classification of the Malvatheca clade (Malvaceae) on the basis of phylogenomics
Gustavo Luna, Lucas Costa, Flávia Fonseca Pezzini, Nisa Karimi, Joeri Sergej Strijk, Jefferson Carvalho-Sobrinho, Matheus Colli-Silva, André Marques, Gustavo Souza

TL;DR
This study explores how hybridization and genome changes have shaped the evolution of the Malvatheca plant clade, revealing complex relationships among its subfamilies.
Contribution
The study provides new phylogenomic evidence of ancient hybridization and polyploidy in the Malvatheca clade, challenging traditional taxonomic classifications.
Findings
Phylogenomic analysis identified four clades within the Malvatheca clade, including a heterogeneous group with mixed subfamily representatives.
Chromosome numbers and repeatome diversity suggest a reticulate evolutionary history, with Bombacoideae showing lower repeat diversity and higher chromosome counts.
Ancient hybridization and polyploidy are shown to be central to the diversification of the Malvatheca clade.
Abstract
Reticulate evolution (RE), involving hybridization and related processes, generates network-like rather than strictly bifurcating relationships among lineages and can obscure phylogenetic relationships. Detecting ancient hybridization is particularly challenging, as genomic signals may erode over time. The Malvatheca clade (Malvaceae), marked by multiple paleopolyploidy events since it’s estimated origin 66 my, offers a useful model for examining RE. Its three subfamilies—Bombacoideae (with high chromosome numbers, mostly trees), Malvoideae (lower chromosome numbers, mostly herbs), and the recently described Matisioideae—show unresolved relationships, with several taxa of uncertain placement. We conducted a phylogenomic analysis of 69 Malvatheca species via complete plastomes, 35S rDNA cistrons, nuclear low copy genes and comparative repeatome data. Most of the datasets consistently…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
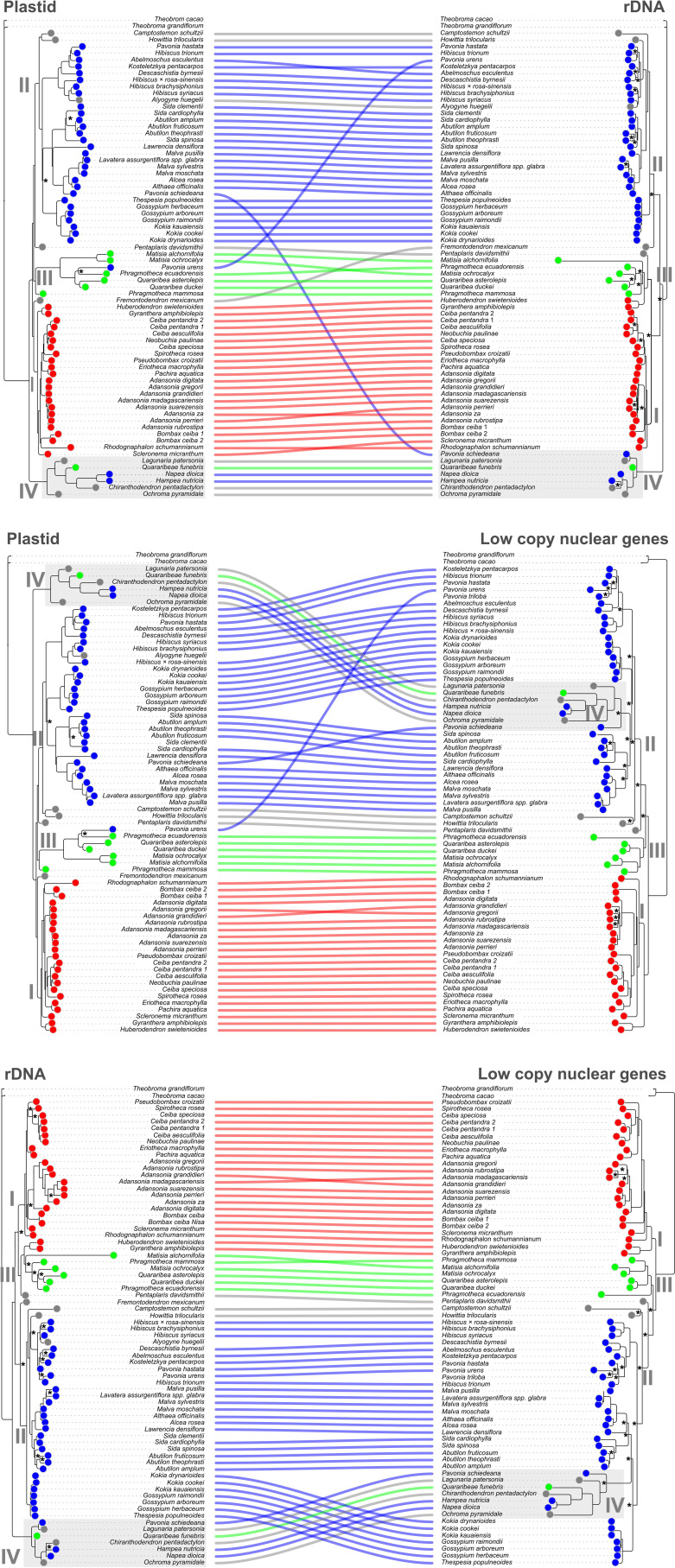 Figure 1
Figure 1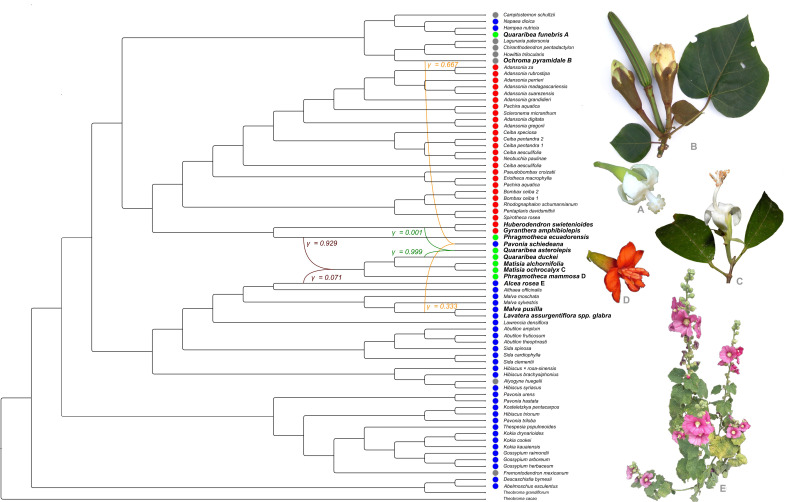 Figure 2
Figure 2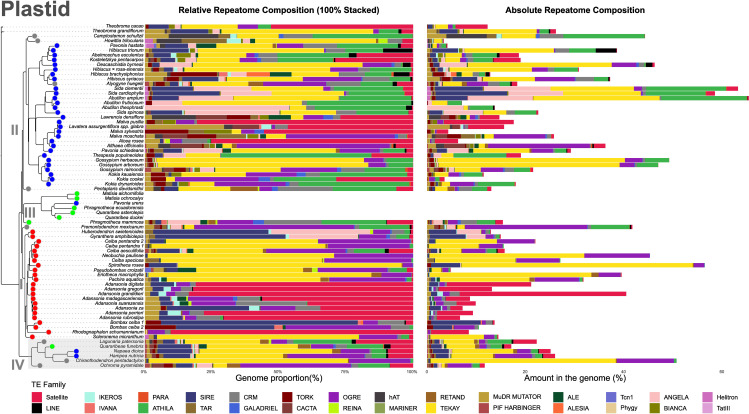 Figure 3
Figure 3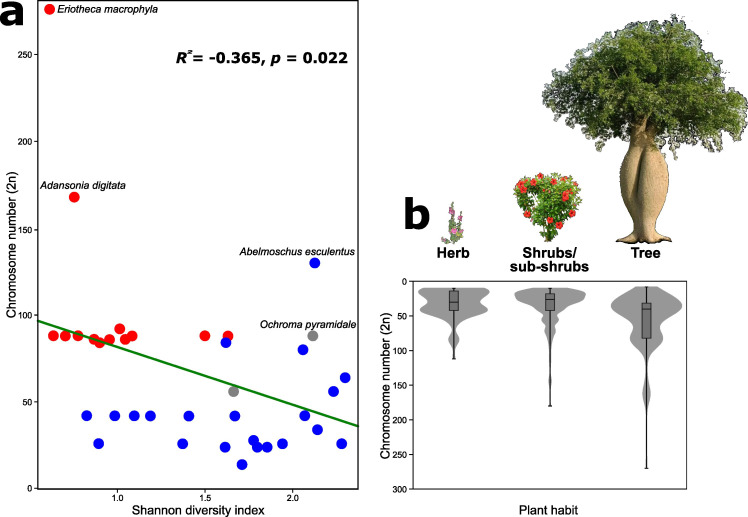 Figure 4
Figure 4| Bombacoideae |
| 1C (PG) | Access | Approach |
|---|---|---|---|---|
| 84 | 1.7 | SRX19282309 | WGS | |
| 44 | 0.76 | SRX24110595 | WGS | |
| 44 | 0.68 | SRX24110598 | WGS | |
| 44 | 0.69 | SRX24110597 | WGS | |
| 42 | 0.71 | SRX7064913 | HybSeq | |
| 44 | 0.65 | SRX24110596 | WGS | |
| 44 | 0.74 | SRX7064911 | HybSeq | |
| 44 | 0.63 | SRX7064903 | HybSeq | |
| 46 | 1.62 | SRX20143449 | WGS | |
| 46 | 1.62 | SRX7064904 | HybSeq | |
| – | – | PRJNA579976 | HybSeq | |
| 43 | 1.75 | SRX20143754 | WGS | |
| 43 | 1.75 |
| HybSeq | |
| 43 | 1.25 | SRX18461125 | WGS | |
| 138 | 4.77 | SRX14481707 | HybSeq | |
| – | – | SRX14481737 | HybSeq | |
| – | – | SRX14481725 | HybSeq | |
| – | – | SRX14481682 | HybSeq | |
| 46 | 2.3 | PRJNA1288544 | HybSeq | |
| – | – | SRX14481702 | HybSeq | |
| – | – | SRX7064905 | HybSeq | |
| – | – | PRJNA579976 | HybSeq | |
| – | – | SRX7064906 | HybSeq | |
| 44 | – | SRX14481726 | HybSeq | |
| Malvoideae | ||||
| 65 | 1.23 | SRX21378022 | WGS | |
| – | – | SRX5462903 | WGS | |
| 21 | – | ERX12138327 | HybSeq | |
| 21 | 1.4 | SRX9130922 | WGS | |
| 21 | – | ERX9653860 | WGS | |
| 21 | – | SRX2855182 | WGS | |
| – | – | ERX7170161 | HybSeq | |
| 13 | 1.7 | SRX5759203 | WGS | |
| 13 | 1.7 | SRX9217821 | WGS | |
| 13 | 0.9 | SRX341462 | WGS | |
| 13 | – | SRX14481653 | HybSeq | |
| – | – | SRX5462993 | WGS | |
| 42 | – | ERX9653895 | WGS | |
| 40 | – | SRX529352 | WGS | |
| 28 | – | SRR17686673 | WGS | |
| – | – | ERX7170167 | Hybseq | |
| 12 | 0.57 | SRX24024016 | WGS | |
| 12 | 0.6 | SRX3304973 | WGS | |
| 12 | 0.57 | SRX24024021 | WGS | |
| 17 | – | SRX12399957 | WGS | |
| – | – | SRX5463054 | WGS | |
| – | – | SRX23987976 | HybSeq | |
| 21 | 1.5 | ERX3149988 | WGS | |
| 21 | 1.4 | ERX3149982 | WGS | |
| 21 | – | ERX5310142 | WGS | |
| 14 | 8.5 | SRX30129087 | WGS | |
| – | – | SRX14481669 | HybSeq | |
| 28 | – | ERX7170168 | HybSeq | |
| – | – | ERR5165833 | HybSeq | |
| – | – | ERR7622263 | HybSeq | |
| – | – | SRX5462742 | WGS | |
| – | – | SRX5462743 | WGS | |
| 7 | 1 | SRX5462745 | WGS | |
| 13 | – | SRX706235 | WGS | |
| Matisioideae | ||||
| – | – | ERR7622266 | HybSeq | |
| – | – | ERR7622272 | HybSeq | |
| – | – | ERR7622267 | HybSeq | |
| – | – | SRX14481713 | HybSeq | |
| – | – | SRR15016217 | HybSeq | |
| – | – | ERR14030174 | HybSeq | |
| – | – | SRX14481652 | HybSeq | |
| Incertae sedis | ||||
| – | – | SRX14481717 | HybSeq | |
| – | – | SRX14481714 | HybSeq | |
| 32 | – | SRX14481721 | HybSeq | |
| – | – | SRX14481650 | HybSeq | |
| – | – | SRX25649159 | HybSeq | |
| 42 | 2.2 | SRX14481665 | HybSeq | |
| Outgroup (Byttnerioideae) | ||||
| 20 | 0.4 | SRX23903616 | WGS | |
| 20 | 0.51 | SRX17564460 | WGS | |
- —Conselho Nacional de Desenvolvimento Científico e Tecnológico10.13039/501100003593
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPlant Diversity and Evolution · Chromosomal and Genetic Variations · Plant Taxonomy and Phylogenetics
Introduction
Reticulate evolution (RE) refers to processes in which genetic material is exchanged between lineages, producing network-like rather than strictly bifurcating phylogenetic relationships (Arnold, 1997; Gontier, 2015). It can arise through hybridization, horizontal gene transfer, or endosymbiosis, and challenges the traditional tree-like view of evolution (Mallet, 2007). Reticulation also complicates classification by blurring taxonomic boundaries and generating conflicting phylogenetic signals. In plants, where hybridization is common, genetic and phenotypic distinctions may be obscured, leading to uncertain or misleading taxonomic boundaries (Mallet, 2007).
Polyploidy (whole genome duplication, WGD), the presence of multiple complete genomes in a species, is closely linked to reticulation and is a recurrent feature of angiosperm evolution (Bowers and Paterson, 2021). Allopolyploidization, i.e., hybridization followed by genome doubling, has been recognized as a major driver of plant diversification (Paterson et al., 2004; Jiao et al., 2011; Vekemans et al., 2012). Notably, a burst of polyploidization events in flowering plants occurred near the Cretaceous–Paleogene (K–Pg) boundary (~66 Mya), coinciding with a mass extinction event (Vanneste et al., 2014; Fawcett and Van de Peer, 2010). The clustering of independent WGDs across angiosperms during this period suggests that polyploidy promoted survival and diversification by providing raw material for genetic novelty (Fawcett et al., 2009; Wood et al., 2009).
Studying RE in ancient lineages remains difficult because signals of hybridization and polyploidy are often eroded over time (Zhang et al., 2025). Additionally, disentangling processes such as incomplete lineage sorting, horizontal transfer, or genetic erosion is challenging (Mallet, 2007), and modeling ancient reticulation requires large datasets and sophisticated analyses. High-throughput sequencing has made these investigations feasible (Dodsworth et al., 2019). Target enrichment methods such as Hyb-Seq enable the analysis of thousands of low-copy nuclear loci (Karbstein et al., 2022), whereas genome skimming can recover plastid genomes (tracing maternal inheritance), ribosomal DNA (biparental nuclear signal), and repetitive elements such as satellites and transposable elements (Dodsworth, 2015; Cavallini et al., 2019). Repeat abundance and sequence similarity offer complementary, alignment-free tools for phylogenetic reconstruction (Dodsworth et al., 2019; Vitales et al., 2020).
The Malvatheca clade (Malvaceae), which originated near the K–Pg boundary (~66 Mya; Hernández-Gutiárrez and Magallon, 2019; Cvetković et al., 2021), serves as a system for investigating these processes. It comprises Bombacoideae (17 genera, ~160 species), Malvoideae (78 genera, ~1,670 species), and the recently recognized Matisioideae (three genera, ~138 species) (Baum et al., 2004; Carvalho-Sobrinho et al., 2016; Cvetković et al., 2021; Colli-Silva et al., 2025). Matisioideae, formerly the tribe Matisieae, was elevated to subfamily rank by Colli-Silva et al. (2025) after consistent recovery of its monophyly, despite its morphological distinctiveness. While Matisioideae is a well-supported lineage, its position—sister to Bombacoideae or to a recircumscribed Malvoideae—remains poorly resolved, even with phylogenomic data. Malvoideae includes widely cultivated herbs such as cotton, hibiscus, and okra (Baum et al., 2004), whereas Bombacoideae consists mainly of tropical trees, including ecologically and culturally significant species such as the baobab (Adansonia L. sp.) and the kapok tree (Ceiba pentandra (L.) Gaertn.) (Wickens, 2008; Zidar and Elisens, 2009). Several genera, such as Ochroma Sw. and Chiranthodendron Larreat., placement remain uncertain due to conflicting morphological and molecular evidence. This phylogenetic uncertainty, coupled with the clade’s history of polyploidy, makes Malvatheca a valuable system for studying reticulate evolution in ancient lineages.
The subfamilies within Malvatheca appear to have followed contrasting genomic trajectories shaped by postpolyploid diploidization. Bombacoideae is predominantly composed of species with high chromosome numbers (2n = 86–276; Costa et al., 2017), whereas Malvoideae ranges from 2n = 10–130, with a modal value near 2n = 16 (Tate et al., 2005). Genomic studies have reported signals of reticulate allopolyploidization in Malvatheca and his consequences to their phylogentic relationships (Hernández-Gutiérrez et al., 2022; Sun et al., 2024; Yang et al., 2025; Zhang et al., 2025), but its consequences for genome architecture, or diversification still poorly known. To address this question, we analyze ribosomal DNA, plastome and repeatome data of 22 Bombacoideae, 34 Malvoideae, 7 Matisioideae, and 6 incertae sedis taxa under a comparative phylogenomic framework. Specifically, we ask the following questions: (i) Is there evidence of reticulate evolution in the group, and is it associated with taxa of uncertain placement? (ii) Are there repeatome signatures linked to ancient WGDs? (iii) Does lifeform correlate with the distinct diploidization pathways observed in Bombacoideae and Malvoideae?
Materials and methods
Data acquisition and repeatome characterization
We analyzed 69 species of the Malvatheca clade, including 34 Malvoideae, 22 Bombacoideae, 7 Matisioideae, and 6 incertae sedis. Two Theobroma species (Bytnerioideae) were included as outgroups. Species names and NCBI accession codes (when available) are listed in Table 1. Target-capture sequencing was used to expand the representation of Bombacoideae, following Costa et al. (2021). Not all species had reads with sufficient reads quality/amount to proceed with all analysis. Thus it was not possible to perform repeatome characterization based on most of the Matisioideae species (except Pragmotheca mammosa and Quararibea funerabis).
Repeatome analyses were performed with the RepeatExplorer2 pipeline implemented in the Galaxy server (https://repeatexplorer-elixir.cerit-sc.cz; Novák et al., 2020), which clusters reads on the basis of sequence similarity. Two approaches were applied: (i) individual analyses, where each species was analyzed separately to characterize its repetitive fraction, and (ii) comparative analysis, where concatenated reads from all species were analyzed jointly to compare repeat composition across lineages.
For individual analyses, ~0.1× genome coverage per species was used (Table 1). For species with unknown genome sizes, values were estimated on the basis of closely related taxa. For the comparative analysis, reads were normalized to ~0.05× per species, yielding a total of 61,288,798 reads. In both cases, clustering was performed using 90% sequence similarity and 55% minimum overlap. The proportions of repeat lineages were calculated as the number of reads per cluster relative to the total number of reads analyzed, excluding chloroplast and mitochondrial sequences identified as potential contaminants.
Correlations between karyotypic, genomic and ecological traits
Following Schley et al. (2022), we treated the genome as a “community” and repetitive element lineages as “species,” allowing the application of community ecology metrics to genome composition. Repeat diversity was quantified via the Shannon diversity index (H; Shannon, 1948), which measures the probability that two randomly chosen repeat copies belong to the same lineage. Higher H values indicate greater repeat diversity.
For each species, diversity indices were calculated from repeat abundances obtained from the RepeatExplorer individual analyses. To test whether repeat diversity was associated with chromosome number, we performed a Pearson correlation between log_10_;-transformed Shannon index values and log chromosome counts across Malvatheca species via the stats package in R (R Core Team, 2019).
To further examine potential ecological correlates, we compiled chromosome numbers and growth habits (herbaceous vs. woody) for 84 species from the literature. Relationships between habit and chromosome number were visualized with boxplots constructed in PAST 4 (Hammer et al., 2001).
Plastome, rDNA, low copy nuclear genes and repeat-based phylogenies
Plastomes and ribosomal DNA (rDNA) sequences were assembled for all sampled species using a reference-based mapping approach in Geneious v6.0.3 (Kearse et al., 2012). Theobroma cacao (NC_014676.2) served as the reference for plastome assembly, while sequence JQ228369.1 was used for rDNA. Reads were mapped to their respective references using the “Map to reference” function, and consensus plastome sequences were aligned with MAFFT (Katoh and Standley, 2013).
The raw reads from each of the 69 species were mapped against 44,343 gene sequences from the Ceiba pentandra genome assembly (Shao et al., 2024) using the “Map to reference” function. These mapped genes were filtered based on mapping quality, phylogenetic resolution, and a coverage threshold requiring the gene to be present in at least 67 species. This filtering process yielded four genes suitable four phylogenetic analysis (CpUnG0025.1, CpUnG0488.1, CpUnG0595.1, CpUnG1740.1). A maximum likelihood (ML) phylogenetic tree was inferred from these four genes, the plastome and the rDNA alignments using IQ-TREE software (v3.0.1). The resulting tree was visualized in FigTree (https://tree.bio.ed.ac.uk/software/figtree/). Topological incongruences between the plastid, rDNA, and nuclear gene trees were visualized using a circular tanglegram generated with the circlize package (Gu et al., 2014) in R. Phylogenetic inference based on repeat abundances was performed following Dodsworth et al. (2015). The abundances of repeat classes obtained from comparative repeatome analyses were treated as quantitative characteristics. Parsimony analyses were conducted in PAST 4 (Hammer et al., 2001).
Reticulate evolution
Reticulate evolution was assessed by constructing phylogenetic networks in PhyloNet 3.8.2 (Wen et al., 2018). The input for this analysis consisted of the individual tree topologies from the complete plastome, rDNA, and four nuclear loci. We employed a two-step approach: (1) estimation of an underlying ML species tree from the set of input gene trees using the DeepCoalCount_tree algorithm (H_0_); and (2) inference of a phylogenetic network by iteratively adding reticulations to this species tree with the DeepCoalCount_network algorithm to estimate the number of hybridization events (H_1_, H_2_, H_3_ etc.). The inferred network was visualized using SplitsTree v6.4.17 (Huson and Bryant, 2006). To independently validate the crosslinking events proposed by network inference, we performed Patterson’s D-statistic test (ABBA-BABA test) based on nucleotide site patterns (Green et al., 2010; Durand et al., 2011). For this, we used information from all loci (plastomes + rDNA + low-copy nuclear genes) analyzed here for representatives of the Malvatheca clade. We calculated the D value for specific taxonomic quartets ((P1, P2), P3, O) designed to test each gene flow hypothesis, where a significant deviation from zero (D > 0) indicates an excess of shared derived alleles between P2 and P3, consistent with introgression.
Results
Comparison between plastidial, rDNA and low-copy nuclear genes topologies
Clade I (Bombacoideae) was well-supported across all three topologies, with one key exception: Huberodendron swietenioides + Gyranthera amphibiolepsis were absent from the rDNA topology, appearing instead within Clade III (Figure 1). Furthermore, on the plastid topology, the incertae sedis species Fremontodendron mexicanum was resolved as the earliest-diverging lineage of this clade (Figure 1).
Phylogenetic relationships within the Malvatheca clade (Malvaceae) inferred from plastome, nuclear rDNA and low copy nuclear genes. Tip labels are color-coded by taxonomic group: Malvoideae (blue), Bombacoideae (red), Matisioideae (green), and incertae sedis (gray). Lines connect the same taxa across the topologies, highlighting congruences and conflicts. Black asterisks stand for support values lower than 70.
Clade II, also well-supported, primarily comprises Malvoideae species. Exceptions include Pavonia urens, which was placed in Clade III (Matisioideae) on the plastid topology, and Pavonia schiedeana, which appeared in Clade IV in the low-copy nuclear and rDNA topologies (Figure 1). This clade also includes the incertae sedis species Camptostemon schultzii, Howittia trilocularis, and Pentaplaris davidsmithii; the latter was placed here in the plastid topology, while Fremontodendron mexicanum was placed here in the rDNA topology (Figure 1).
Clade III (Matisioideae) was consistently well-supported (Figure 1). However, several species from other groups appeared within it depending on the topology: the incertae sedis species Pentaplaris davidsmithii and the Bombacoideae species H. swietenioides and G. amphibiolepsis on the rDNA topology; the Malvoideae species Pavonia urens on the plastid topology (Figure 1). On the other hand, the Matisioideae specie Quararibea funebris was placed in another clade (IV) across all analyses (Figure 1). Remarkable, the backbone of the subfamilies was variable depending on the dataset, with clade III (Matisioideae) being sister to Bombacoideae in rDNA topology or sister to Malvoideae in plastidial and low-copy nuclear gene topologies (Figure 1).
Clade IV is morphologically unclear composed of three incertae sedis species (Chiranthodendron pentadactylon, Lagunaria patersoniana and Ochroma pyramidale), one Matisioideae (Q. funebris), and two Malvoideae species (Hampea nutricia and Napea dioica). Additionally Pavonia schiedeana was an exception, appearing in this clade only in the low-copy nuclear and rDNA topologies, not in the plastid topology (Figure 1). In the low copy nuclear genes topology, these species were placed within the Clade II. The incongruence and relationship details are present in Supplementary Figures 1, 2.
Reticulate evolution in Malvatheca
By assuming positional changes in nuclear and plastidial topologies as evidence of reticulate evolution, our data revealed a high complexity among plastidial, rDNA, and low-copy nuclear gene topologies (Figure 1). Major topological incongruences involved the placement of two Pavonia species. P. urens was resolved within Clade III in the plastid topology, while P. schiedeana was placed in Clade IV in both the low-copy nuclear and rDNA topologies. The PhyloNet analysis provided strong evidence for reticulate evolution in Malvatheca. The DeepCoalCount function perform a species tree with a ‘total number of extra lineages score’ of 695.0. Subsequent testing the distinct reticulation hypotheses (H_1_–H_4_) using the DeepCoalCount_network algorithm yielded the following scores: H_1_ = 517.0, H_2_ = 497.0, H_3_ = 456.0, and H_4_ = 466.0 (The lower this score, the less conflict there is between the tested loci). Thus, H3 was selected and this network reveal three statistically supported reticulation events within the Malvatheca clade (Figure 2): (i) A hybrid origin of Pavonia schiedeana Steud. involving the cross between the lineages of Malva pusilla Sm. + Lavatera assurgentiflora subsp. glabra (Cockerell) Guilliams clade (inheritance proportion, γ = 0.333) and Ochroma pyramidale (Cav. ex Lam.) Urb. (γ = 0.667); (ii) a hybrid origin of Quararibea asterolepis Pittier by the cross between the lineages Huberodendron swietenioides (Gleason) Ducke + Gyranthera amphibiolepis W.Palacios clade (γ = 0.001) and Quararibea duckei Huber (γ = 0.999); (iii) a ancient hybridization between ancestors lineages of Phragmotheca ecuadorensis W.S.Alverson (γ = 0.929) and Alcea rosea L. (γ = 0.071), which gave rise to the entire clade III (Figure 2). Network inference (PhyloNet) under Maximum Parsimony and Maximum Likelihood criteria rejected strict bifurcation in favor of a network with three hybridization events. We independently validated the two main events using D-statistics (ABBA-BABA test) based on nucleotide site patterns. We detected strong evidence of introgression between the Pavonia lineage and the Malveae clade (D = 0.61), as well as between the tribe Matisieae (Quararibea) and the genus Huberodendron (D = 0.54), confirming that the observed topological discordance is not merely the result of incomplete lineage sorting (ILS), but rather of significant ancient gene flow. On the other hand, the reticulation at the base of the Matisioideae clade showed D = 0.05, with the distribution of shared (ABBA) and unshared (BABA) alleles being almost perfectly symmetrical (19 to 17), suggesting a scenario of Incomplete Lineage Sorting (ILS).
The optimum phylogenetic network inferred from single gene trees of the four nuclear low copy genes + rDNA + plastomes performed for Malvatheca clade (Malvaceae) using PhyloNet analyses.
Repeatome diversity in the Malvatheca clade
In the individual repeatome analyses, the sequencing depth ranged from 40,572 reads in Bombax ceiba (accession 2) to 2,633,822 in Hibiscus syriacus (Supplementary Table 1). The proportion of repetitive DNA varied widely, from 4.8% in Pseudobombax croizatii A. Robyns to 56.4% in Spirotheca rosea (Seem.) P.E.Gibbs & W.S. Alverson (Bombacoideae), and from 3.4% in Howittia trilocularis to 65.4% in Abutilon amplum Benth. (Malvoideae) (Figure 3; Supplementary Table 2).
Repeatome composition of the Malvatheca clade. The plastome-based phylogeny (left) is shown alongside the repeat profiles for each species (right). Stacked bars indicate the relative proportions of major repeat classes (normalized to 100%), whereas dots represent their estimated absolute genomic abundance (Mb).
Both subfamilies were dominated by Class I transposable elements, particularly LTR retrotransposons of the Ty3-type superfamily. Their abundance ranged from 0.5% in Malva sylvestris L. to 50% in Chiranthodendron pentadactylon Larreat. (Supplementary Table 2). Within Bombacoideae, Tekay elements predominated across four genera, whereas Ogre elements also occurred at moderate to high levels. In Malvoideae, Tekay and Ogre were generally present in moderate proportions, with a few species showing elevated abundances of Athila elements. Among Ty1-copia elements, Angela and SIRE were the most widespread across both subfamilies, with SIRE being particularly enriched in Sida cardiophylla Domin. and one Bombax ceiba accession (Figure 3; Supplementary Table 3). The satellite DNA content also varied considerably, from 0.02% in Gossypium arboreum L. to 33.9% in Adansonia grandidieri Baill. Bombacoideae, especially Adansonia, tended to have relatively high satellite DNA abundances (4.9–33.8%). Despite this variation, no genomic synapomorphies based on repeat presence/absence were detected that clearly distinguished Bombacoideae from Malvoideae (Figure 3; Supplementary Tables 2, 3).
Comparative repeatome analysis revealed limited sharing of repetitive DNA clusters between subfamilies. Within Bombacoideae, intrageneric similarity was high: Adansonia species shared the most repeat clusters, and Pseudobombax croizatii and Pachira aquatica Aubl. Both genera presented elevated proportions of Ty3-type Ogre and CRMs. In contrast, Malvoideae species presented highly divergent repeatomes, with even congeneric taxa (e.g., Malva sylvestris and M. moschata L.) sharing few clusters (Supplementary Figure 3; Supplementary Table 4).
The repeat-based phylogeny recovered three major clades but with generally weak support (Supplementary Figure 4; Supplementary Table 4). Clade III showed extensive mixing, grouping representatives of all subfamilies without resolution. Several genera were not recovered as monophyletic, including Adansonia (Bombacoideae) and Abutilon, Sida, Hibiscus, and Malva (Malvoideae). Three Bombacoideae species (Phragmotheca mammosa, Gyranthera amphibiolepis W. Palacios, Huberodendron swietenioides (Gleason) Ducke) and four Malvoideae species (Camptostemon schultzii Mast., Pentaplaris davidsmithii Dorr & C. Bayer, Alyogyne huegelii (Endl.) Fryxell, Pavonia schiedeana Steud.) shifted into clade III. Additional rearrangements, such as altered relationships among Adansonia, Scleronema Benth, and Rhodognaphalon (Ulbr.) Roberty (clade I) and the placement of Pavonia, Decaschistia, and two Hibiscus species in clade II (Supplementary Figure 4; Supplementary Table 4).
Comparison with the rDNA tree revealed similar patterns: multiple species from clades I and II were grouped into clade III, with further positional changes within clades (e.g., Bombax ceiba, Fremontodendron, and Rhodognaphalon in clade I; Howittia, Hibiscus, and Decaschistia byrnesii Fryxell in clade II) (Supplementary Figure 4; Supplementary Table 4).
Diversity of repeats and correlation with chromosome number
The repeat diversity of Malvatheca, as measured by the Shannon (H) and Simpson (D) indices, ranged from very low values in Rhodognaphalon schumannianum (A. Robyns) A. Robyns (H = 0.4, D = 0.17) to high values in Hibiscus brachysiphonius F. Muell. (H = 2.4, D = 0.9). Malvoideae species generally presented greater repeat diversity (H = 0.8–2.4; D = 0.3–0.9) than did Bombacoideae, which tended toward lower values (Figure 4a; Supplementary Table 5). Chromosome number was negatively correlated with repeat diversity (Shannon: R = –0.36, p = 0.022; Simpson: R = –0.39, p = 0.011), indicating that species with higher chromosome counts, such as Bombacoideae, typically presented fewer diverse repeatomes, whereas Malvoideae (lower 2n) presented greater diversity (Figure 4b; Supplementary Table 5).
Repeat diversity and chromosome number variation in Malvaceae. (a) Shannon diversity index of repetitive DNA elements in Malvatheca species, colored by taxonomic group: Bombacoideae (red), Malvoideae (blue), and incertae sedis (gray). (b) Chromosome number distributions across growth habits (herbs, shrubs/subshrubs, and trees) compiled from Malvaceae species.
To assess whether chromosome number is correlated with growth habits, we compiled data from 84 Malvaceae species. Trees presented consistently greater chromosome numbers than shrubs and herbs did (range: 8–328; median = 30), supporting an association between the arboreal habit and large chromosome complements (Figure 4b; Supplementary Table 6).
Discussion
Would the diversification of Malvatheca be driven by reticulate evolution?
Our data provides phylogenetic evidence for RE in the Malvatheca clade, corroborating previous genomic analyses (Karimi et al., 2020; Wan et al., 2024; Zhang et al., 2025). These reticulation events appear to have occurred on different timescales (Hernández-Gutiérrez et al., 2022; Yang et al., 2025; Stull et al., 2023), ranging from more recent lineages (e.g., the 21.6 Mya genus Adansonia; Wan et al., 2024) to the 53.5 Mya origin of Bombacoideae (Zizka et al., 2020). Hybridization has long been recognized as a fundamental evolutionary force shaping plant diversity, with impacts ranging from immediate reproductive isolation to long-term adaptive radiation (Rieseberg, 1997; Soltis et al., 2004; Chester et al., 2012). Phylogenetic analysis of Malvaceae revealed a complex reticulated evolutionary history. Network inference (PhyloNet) under Maximum Parsimony and Maximum Likelihood criteria rejected strict bifurcation in favor of a network with three hybridization events. The clades III and IV were consistently associated with reticulation points in both plastid and nuclear datasets, suggesting a complex hybrid origin (Hernández-Gutiérrez et al., 2022; Yang et al., 2025). The morphological and cytogenetic heterogeneity of its members supports this view. Sampling the only three genera of Matisioideae (Matisia, Quararibea, and Phragmotheca) allowed us to demonstrate that most representatives of the subfamily (except Q. funebris) are grouped into a well-supported clade. The hybrid origin test of this clade, involving a reticulation event between Bombacoideae and Malvoideae, was inconclusive, and our analyses suggest that it may simply be a case of high levels of deep incomplete lineage sorting (ILS) among these subfamilies. Regarding the methods used here to calculate hybridization or introgression (D-statistics), it is worth noting that these tests can be highly sensitive to evolutionary rate variation across different lineages, which can frequently lead to false positives or inconclusive results (Frankel and Ané, 2023). Given the phylogenetic, morphological, and systematic complexity of the relationship between the Malvaceae subfamilies (Colli-Silva et al., 2025), these hypotheses of reticulated evolution need to be tested further with a larger sample size. There are no widely sampled phylogenetic hypotheses for the genus Quararibea; therefore, the unexpected position of Q. funebris would need to be investigated in more detail, and may be related to taxonomic identification error, reticulation, and/or methodological limitations (e.g., low coverage).
While nuclear low copy genes, rDNA and plastid datasets provided strong phylogenetic resolution for Malvatheca, the repeat-based approach (Dodsworth et al., 2015) failed to resolve relationships. This loss of signal may reflect (i) the impact of reticulation, as hybridization, particularly in allopolyploid contexts, can induce substantial restructuring of repetitive elements through sequence loss, differential retention of parental repeats, and epigenetic reorganization, thereby altering repeat presence, absence, and relative abundance (Parisod et al., 2010), and/or (ii) the deep divergence of Bombacoideae (53.5 Mya; Zizka et al., 2020). Because repeats undergo rapid evolutionary turnover, their phylogenetic utility can erode in ancient groups. Nevertheless, recent studies have shown that repeatome composition can retain signals and even capture ancient hybridization through characteristic proliferation patterns (Castro et al., 2024; Hlavatá et al., 2024). The contrast across plant groups highlights the lineage- and timescale-specific nature of repeat data. For example, in Erythrostemon Klotzsch (Leguminosae, 33.6 Mya), systematic variation in repeat composition revealed discordant profiles consistent with ancient origins (Castro et al., 2024). Similarly, in Amomum L. (Zingiberaceae, 19.3 Mya), bursts of repeat proliferation coincided with inferred hybridization events, driving genome size changes that track clade divergence (Li et al., 2023; Hlavatá et al., 2024).
Genomic-scale phylogenies further demonstrated that reticulate evolution is pervasive across Malvaceae, challenging tree-based models (Hernández-Gutiérrez et al., 2022; Yang et al., 2025; Zhang et al., 2025). Using 268 nuclear loci across 96 genera, Hernández-Gutiárrez and Magallon (2019) detected extensive discordance attributable to incomplete lineage sorting and introgression, showing that bifurcating trees fail to capture the family’s evolutionary complexity. Reticulation manifests at multiple scales, from recent interspecific gene flow to ancient allopolyploid events. Hyb-Seq datasets and network inference have revealed well-supported introgression in baobabs (Adansonia), clarifying floral homoplasy and pollination biology (Karimi et al., 2020). Similarly, independent hybridization and introgression across Gossypium highlight the role of allopolyploidy in diversification (Zhang et al., 2025). Cytonuclear processes in Hibiscus (Pfeil, 2002) involve chloroplast capture, whereas cytogenetic work in Eriotheca (Serra et al., 2022) confirms that hybridization and polyploidy remain active processes. Together, these findings underscore that phylogenetic networks, rather than bifurcated trees, more accurately represent Malvaceae evolution and highlight the central role of RE in shaping its genomic and phenotypic diversity.
The repeatome compositions of the Malvatheca clade subfamilies
Genomic studies indicate that the Malvatheca clade has undergone multiple rounds of polyploidization, including reticulate allopolyploidy and subsequent dysploidy, which have played central roles in its diversification (Hernández-Gutiérrez et al., 2022; Yang et al., 2025; Zhang et al., 2025). Recent chromosome-scale assemblies of Adansonia spp., Bombax ceiba and Ceiba pentandra have provided unprecedented insights into genome structure and repeat composition across Malvaceae (Wan et al., 2024; Shao et al., 2024). Complementary phylogenomic studies, such as those on Hibiscus L., have revealed at least three independent whole-genome duplication (WGD) events (Eriksson et al., 2021), highlighting the recurrent nature of polyploidy within the family. These duplications supplied raw genetic material for evolutionary novelty, whereas subsequent post-polyploid diploidization processes reshaped karyotypes and contributed to current genomic diversity patterns.
The broader evolutionary significance of WGD has been well documented across angiosperms. Large-scale comparative studies have shown that WGDs are not randomly distributed in time but cluster during episodes of environmental stress (Van de Peer et al., 2017; Teng et al., 2022). This has given rise to the so-called “polyploidy paradox”: although WGD frequently occurs, only a subset of polyploid lineages survive in the long term (Van de Peer et al., 2021). Several of these surviving events appear to coincide with the Cretaceous–Paleogene (K–Pg) boundary (~66 Mya), a period of mass extinction. WGDs at this time are hypothesized to have facilitated lineage survival and diversification by buffering against genomic stress and providing redundancy for adaptive innovation (Fawcett et al., 2009; Baduel et al., 2018; Bomblies, 2020). The “polyploid hop” model further proposes that polyploidy creates both challenges and opportunities: initial barriers to establishment may be overcome by long-term adaptive potential (Baduel et al., 2018). Recent studies also suggest that specific life-history traits, such as seed biology, may interact with polyploid status to influence which lineages persist across the K–Pg boundary (Cai et al., 2019; Berry and Jaganathan, 2022).
Within Malvaceae, repeatome dynamics provide important clues to post-WGD genome evolution. Chromosome-scale assemblies have shown that transposable element (TE) proliferation and chromosome restructuring are major forces shaping genome size and karyotype variation (Li et al., 2024; Shao et al., 2024). In Malvatheca, distinct repeatome profiles are observed among subfamilies: Bombacoideae tends to exhibit lower repeat diversity, Malvoideae has higher abundance and diversity, and Matisioideae has intermediate patterns. These differences likely reflect contrasting evolutionary strategies for restructuring genomes after polyploidy, with Bombacoideae favoring karyotypic stability through TE amplification, Malvoideae accumulating a more diverse repeat fraction, and Matisioideae retaining a mixed profile.
Taken together, these patterns suggest that the interplay among ancient WGDs, repeatome evolution, and post-polyploid diploidization has been a major driver of lineage-specific diversification in Malvatheca. Variation in repeat composition and chromosome number across subfamilies reflects different genomic strategies for managing the legacy of polyploidy, highlighting the central role of repetitive DNA in shaping both the stability and flexibility of plant genomes over deep evolutionary timescales.
The habit correlated with chromosome number
Our results revealed that tree-dominated lineages presented the highest chromosome numbers. In Bombacoideae, striking examples include Pseudobombax and Pachira (2n = 88–92), Adansonia digitata (2n = 160), and Eriotheca species, with exceptionally elevated counts of 2n = 194–276 (Costa et al., 2017). A similar pattern was described for incertae sedis, such as Ochroma pyramidale (2n = 84) (Costa et al., 2017). A similar trend occurs in the exclusively arboreal Tilioideae, where counts range from 2n = 82 to 2n = 328 (Pigott, 2002; 2012). These data reflect the trend toward ancient whole-genome duplication (WGD) events, followed by the lineage-specific conservation of high chromosome numbers in long-life-cycle species.
Shrubby lineages, such as Hibiscus (2n ≈ 52) and Pavonia (2n ≈ 56), present intermediate values, possibly representing transitional stages between herbaceous and woody forms. In contrast, herbaceous genera such as Sida are chromosomally conserved, typically ranging from 2n = 14–32 (Skovsted, 1935; Fernández et al., 2003). These reductions likely result from dysploidy — the step-wise loss or fusion of chromosomes — a process often associated with short-lived, fast-reproducing life cycles (Siljak-Yakovlev et al., 2017).
The habit–chromosome relationship thus reflects contrasting evolutionary pressures. In woody clades such as Bombacoideae, ancient polyploidization established high baselines that were further expanded by more recent polyploidy (Marinho et al., 2014). Herbaceous lineages, in turn, underwent systematic reductions, which is consistent with patterns observed in other families, such as Asteraceae, where perennial-to-annual transitions are linked to smaller karyotypes (Watanabe et al., 1999). Selective pressures on herbaceous species — favoring rapid cell cycles and short generation times — likely reinforced this reduction. Similar correlations between life form and karyotype have been documented across angiosperms (e.g., Malpighiaceae, Lombello and Forni-Martins, 2003; Ehrendorfer, 1989), but Malvaceae provides one of the clearest examples: Bombacoideae, dominated by trees, consistently carries higher chromosome numbers than Malvoideae, which include mostly herbs and shrubs. These results suggest that major life form transitions are accompanied by fundamental changes in genome organization.
Conclusions
Our phylogenomic analyses provide strong evidence for reticulate evolution in the Malvatheca clade (Malvaceae), underscoring its role as a fundamental driver of diversification in the family. Ancient hybridization events, coupled with allopolyploidy and subsequent diploidization, have left lasting imprints on the genomic architecture and subfamily relationships. Nuclear and plastid datasets revealed extensive reticulation, whereas repeatome analyses proved less effective for deep phylogenetic resolution, reflecting rapid lineage-specific turnover. This contrast highlights the importance of multilayered genomic approaches for reconstructing complex evolutionary histories. Comparative genomic and cytogenetic evidence also reveals subfamily specific patterns in karyotype evolution. Tree-dominated lineages, particularly Bombacoideae, exhibit exceptionally high and variable chromosome numbers arising from successive polyploidization, whereas herbaceous taxa display reduced counts through dysploidy and genome downsizing. These correlations suggest a close interplay between life form, polyploidy, and genome structure: woody lineages preserve high baselines established by ancient WGDs, whereas herbaceous taxa evolve toward reduced karyotypes under different life-history constraints. Taken together, our results demonstrate how hybridization, polyploidy, and repeat dynamics have jointly shaped Malvaceae diversification. This family offers a compelling model for understanding the integration of reticulate processes, structural genome evolution, and life-history strategies in driving morphological and ecological innovation. Future research incorporating chromosome-scale assemblies and functional studies will be critical for disentangling the genomic consequences of WGD and reticulation across this diverse plant lineage.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Arnold M. L. (1997). Natural Hybridization and Evolution (Oxford: Oxford University Press).
- 2Baduel P. Bray S. Vallejo-Marín M. KolářF. Yant L. (2018). The “Polyploid Hop”: shifting challenges and opportunities over the evolutionary lifespan of genome duplications. Front. Ecol. Evol. 6, 117. doi: 10.3389/fevo.2018.00117 · doi ↗
- 3Baum D. A. De Witt Smith S. Yen A. Alverson W. S. Nyffeler R. Whitlock B. A. . (2004). Phylogenetic relationships of Malvatheca (Bombacoideae and Malvoideae; Malvaceae sensu lato) as inferred from plastid DNA sequences. Am. J. Bot. 91, 1863–1871. doi: 10.3732/ajb.91.11.1863, PMID: 21652333 · doi ↗ · pubmed ↗
- 4Berry K. Jaganathan G. K. (2022). Did selection for seed traits across the Cretaceous/Paleogene boundary sort plants based on ploidy? Acta Palaeobot. 62, 182–195. doi: 10.35535/acpa-2022-0012 · doi ↗
- 5Bomblies K. (2020). When everything changes at once: finding a new normal after genome duplication. Proc. R. Soc B. 287, 20202154. doi: 10.1098/rspb.2020.2154, PMID: 33203329 PMC 7739491 · doi ↗ · pubmed ↗
- 6Bowers J. E. Paterson A. H. (2021). Chromosome number is key to longevity of polyploid lineages. New Phytol. 231, 19–28. doi: 10.1111/nph.17361, PMID: 33772797 · doi ↗ · pubmed ↗
- 7Cai L. Xi Z. Amorim A. M. Sugumaran M. Rest J. S. Liu L. . (2019). Widespread ancient whole-genome duplications in Malpighiales coincide with Eocene global climatic upheaval. New Phytol. 221, 565–576. doi: 10.1111/nph.15357, PMID: 30030969 PMC 6265113 · doi ↗ · pubmed ↗
- 8Carvalho-Sobrinho J. G. Alverson W. S. Alcantara S. Queiroz L. P. Mota A. C. Baum D. A. (2016). Revisiting the phylogeny of Bombacoideae (Malvaceae): Novel relationships, morphologically cohesive clades, and a new tribal classification based on multilocus phylogenetic analyses. Mol. Phylogenet. Evol. 101, 56–74. doi: 10.1016/j.ympev.2016.05.006, PMID: 27154210 · doi ↗ · pubmed ↗