Concept of neuroendocrine neoplasms of all organs with a focus on grading, subtyping
Atsuko Kasajima, Aurel Perren, Günter Klöppel

TL;DR
This paper discusses neuroendocrine neoplasms, focusing on how they are classified, graded, and subtyped based on their biology and clinical features.
Contribution
The paper highlights recent advances in molecular subtyping and grading systems for neuroendocrine neoplasms, emphasizing their clinical and prognostic implications.
Findings
NETs and NECs differ in histology, clinical behavior, and molecular profiles, with NETs often linked to hereditary syndromes.
TP53 mutations and RB1 inactivation are key in NECs, while NETs show mutations in genes like MEN1 and ATRX.
Integrated histological, molecular, and clinical approaches improve classification and management of NEN subtypes.
Abstract
Neuroendocrine neoplasms (NENs) are a heterogeneous group of neoplasms encompassing both well differentiate neuroendocrine tumors (NETs), and poorly differentiated neuroendocrine carcinomas (NECs). This classification is supported by distinct histological, clinical, and molecular profiles. NETs are typically slow-growing and hormone-producing, with organoid architecture and frequent associations with hereditary syndromes such as multiple endocrine neoplasia type 1 (MEN1) and von Hippel-Lindau (VHL) disease. In contrast, NECs are highly malignant, rapidly proliferating tumors characterized by mutations in adenocarcinoma-driver genes and in addition to TP53 mutations and RB1 inactivation, without hereditary links to endocrine tumor syndomes. Recent WHO classifications introduced site-specific grading systems, including NET G3 in the digestive, urogenital, gynecological and head and neck…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
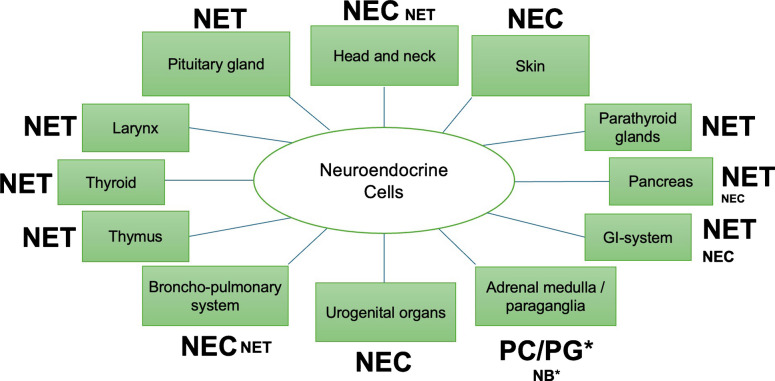 Figure 1
Figure 1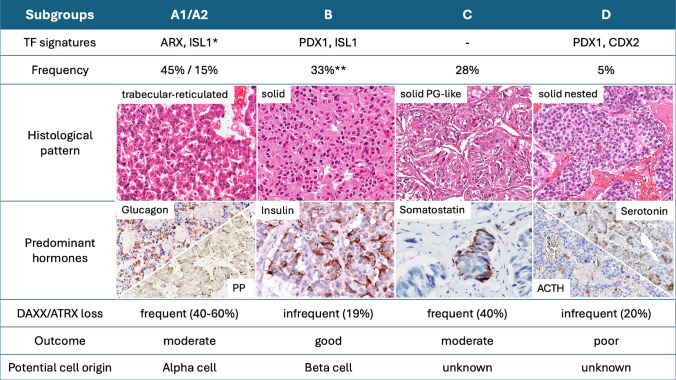 Figure 2
Figure 2- —Technische Universität München (1025)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsNeuroendocrine Tumor Research Advances · Thyroid Cancer Diagnosis and Treatment · Salivary Gland Tumors Diagnosis and Treatment
Introduction
Neuroendocrine neoplasms (NENs) are a group of tumors that appear uniform but are diverse. They can arise in various anatomical sites and are defined by neuroendocrine markers and hormonal secretory granules. Many also express somatostatin receptor 2 (SSTR2).
Despite the prevalence of these common features, NENs exhibit significant variations in morphology, proliferation rate, hormone production, molecular profile, and clinical course. Consequently, classification systems vary depending on differentiation, the organ or tissue of origin and proliferation.
NENs of the digestive and pulmonary systems account for approximately 70% of all NENs and have been pivotal to classification efforts. Both systems adopted three-tiered grading systems that distinguish low-grade (TC), intermediate-grade (AC), and high-grade neoplasms (large cell neuroendocrine carcinoma [LCNEC] and small cell lung carcinoma [SCLC]) for pulmonary NENs [1], and G1, G2 neuroendocrine tumors (NETs) and G3 neuroendocrine carcinomas (NEC) for digestive NENs [2]. The updated grading approach for the digestive system is currently being applied to NENs in other organs, including those in the urogenital and head and neck regions [3]. There is ongoing discourse regarding adapting this approach to NENs in the breast and other sites [4].
NENs were first described in the late nineteenth century in the ileum. The term "carcinoid" was coined by Siegfried Oberndorfer. By the 1930s, the equivalents to carcinoids had been identified in various organs, including the lung, ovary, and gastrointestinal tract. These tumors stained with silver impregnation methods [5, 6]. In 1963, Williams and Sandler classified carcinoids by embryologic origin [7] into foregut, midgut and hindgut tumors. In the WHO classification of 1980, the pancreatic tumors were classified under the term of islet cell adenomas and islet cell carcinomas. It was emphasized that all carcinoids should be regarded as malignant [8]. Also included were poorly differentiated lung carcinomas, called oat-cell carcinomas. The 1980 WHO classification includes carcinoids and undifferentiated neuroendocrine carcinoma as the two primary types of NENs [8]. In 2000, the second edition of the WHO classification for endocrine tumors replaced the terms carcinoid by "well-differentiated endocrine tumor" and "well-differentiated endocrine carcinoma" [9]. This was based on a classification proposal formulated in an article by Capella [10]. The 1999 WHO classification for lung and thoracic organs, however, continued to use the term carcinoid and divided into two categories: typical and atypical. Small cell lung carcinoma (SCLC) and large cell neuroendocrine carcinoma (LCNEC) were classified as undifferentiated neuroendocrine neoplasms of the lung. This classification was retained in all subsequent editions published in 2004, 2015, and 2021 [1, 11, 12].
NENs are a category of neoplasms that manifest as epithelial tumors. These neoplasms are characterized by vesicular granules containing peptide hormones and biogenic proteins with endocrine or paracrine effects and chromogranins in large dense-core vesicles, as well as membrane proteins like synaptophysin in synaptic-like granules containing small mediators (taurine, choline, GABA, etc.). A new endocrine marker is the transcription factor INSM1, which resides in the nucleus of neuroendocrine cells. Neuroendocrine neoplasms with neuroectodermal origin and without cytokeratin expression can be considered a special type among NENs, as their other characteristics largely correspond to those of cytokeratin-positive NENs. As the cells derived from the neuroectoderm colonize the paraganglia (including adrenal medulla) and olfactory membrane, the cells with epithelial features predominantly manifest in the gastrointestinal tract, bronchial system, thyroid (as C-cells), parathyroid glands, and pancreatic islets [13].
Dichotomy of NENs
As knowledge of NENs has increased, it has become increasingly evident that there are substantial similarities among NENs from different anatomical locations, and that these neoplasms can be categorized into two families. This prompted the proposal of a comprehensive classification system that encompasses all NENs and divides them into well-differentiated NENs, called NETs, and poorly differentiated NENs, called NECs [14].
NETs exhibit distinguishing characteristics when compared to NECs in terms of organoid histology, typically exhibiting low proliferation rates and manifesting as indolent, slowly growing tumors. Furthermore, NETs have been linked to hormone production and are associated with hereditary endocrine tumor syndromes, including multiple endocrine neoplasia (MEN1 and MEN2) and von Hippel-Lindau syndrome (VHL) and less frequently Neurofibromatosis type 1 (NF1) and rare endocrine tumor syndromes (see below). Conversely, NECs are aggressive, fast-growing neoplasms that generally do not express hormones or produce hormonal syndromes. NECs are not associated with familial endocrine tumor syndromes. Recent genetic findings provide further evidence that lends support to the existence of the dichotomy of NENs [15–20]. Within the digestive and bronchopulmonary system, NECs frequently harbor TP53 mutations, often accompanied by RB1 inactivation [15–21]. These mutations are only rarely found in digestive and lung NETs [17, 21], where in a site dependent manner mutations in genes such as MEN1, DAXX/ATRX prevail [17, 21]. Another molecular marker is SSTR2, which is frequently found in NETs, and only rarely expressed in NECs [17].
NETs are predominantly observed in the thymus, stomach, small bowel, appendix, rectum, and pancreas. Conversely, NECs exhibit a higher prevalence in the lung, esophagus, colon, and urogenital organs [22]. In certain organs, such as the pituitary gland or skin, the prevalence of NETs or NECs is so low that the WHO classification delineates only a single tumor type. This assertion pertains to both pituitary and parathyroid tumors, wherein the observed neoplasms align with the NET category, even if they are called carcinomas. In the thyroid gland, medullary thyroid carcinoma manifests feature consistent with NET, rather than NEC, the existence of which in the thyroid gland is a subject of debate. The majority of NENs in the skin are Merkel cell carcinomas, corresponding to NECs. Conversely, NETs of the skin are exceedingly rare (Fig. 1).Fig. 1. Distribution in the body and site specific prevalence estimation of neuroendocrine tumors (NETs) and neuroendocrine carcinomas (NECs). Large letters indicate high prevalence, small letters low prevalence. Abbreviation: PC pheochromocytoma, PG paraganglioma, NB neuroblastoma, GI gastrointestinal. *In the adrenal medulla and paraganglia, pheochromocytomas and paragangliomas may be regarded as the cytokeratin negative counterparts of NETs in other regions, given their good differentiation, indolent biological behavior, and relatively frequent association with familial and hereditary tumor syndromes such as MEN2 or VHL. In contrast, neuroblastoma of the adrenal medulla, based on its biological and clinical behavior, may be considered the equivalent of NEC
A critical question that emerges from the analysis of the incidence figures for different NEN types is whether these variations are attributable to different causes. In sporadic NETs, the etiology is not well understood, with the exception of cases associated with germ line gene mutations underlying familial and hereditary tumor syndromes, such as MEN1 or VHL. In contrast, the etiology of NECs is well-defined. A substantial correlation has been identified between smoking and SCLCs, with an association also extending to urinary bladder NECs. A notable proportion of Merkel cell carcinomas have been linked to polyomavirusinfection or ultraviolet irradiation. The development of digestive NECs may be influenced by environmental factors. These neoplasms predominantly manifest in the stomach and colon, regions where extrinsic carcinogens are believed to play a pivotal role in the etiology of the disease. Conversely, the presence of NETs is most notable in gastrointestinal regions, such as the small intestine, where environmental factors appear to exert a relatively minor influence on tumorigenesis. In the adrenal medulla and the paraganglia, the pheochromocytoma and paragangliomas may be considered the equivalent to the NETs in other regions, because of their good differentiation and relatively frequent association with familial and hereditary tumor syndromes, such as MEN2 or VHL, while neuroblastomas of the adrenal medulla correspond to NECs. Tumors of the adrenal cortex do not fit into this concept because they lack essential characteristics of NENs, namely peptide hormone production and hormone granule formation.
Classification and grading
The 2010 WHO classification for NENs of digestive organs introduced a novel system for categorizing NENs. Well-differentiated NENs were designated as NETs and subsequently classified into NET G1 and NET G2 based on their proliferative activity, as determined by Ki-67 index and mitotic counts. NECs were assigned to a G3 status, defined by a Ki-67 index > 20% or mitotic counts > 20 per 2 mm^2^, and subsequently divided into small cell and large cell types [2]. According to the 2017 WHO classification of the endocrine pancreas, the NET grading system was expanded to encompass a group of NET G3 cases that were characterized by a high Ki-67 index (defined as > 20% positive staining) and/or mitotic counts (defined as > 20 mitoses per 2 mm^2^) [23]. In the 2019 WHO classification of digestive system NENs, this three-tiered grading system, designated as G1, G2, and G3, was applied to all NENs within the digestive system, including those located in the hepatobiliary organs (see Table 1) [24]. Recently, a proposal has been made to expand the classification of PanNETs to encompass four distinct grades: G1, G2a, G2b, and G3. Dividing G2 PanNETs into G2a (Ki67 3–10%) and G2b (10%—≤ 20%) provides substantial additional prognostic information for these tumors, as evidenced by a large series of PanNETs [25]. Similar observations were made in studies with a smaller number of patients [26, 27]. Although Ki67 assessment is subject to interobserver variability and methodological differences, standardized protocols can improve consistency. Therefore, the precise Ki67 percentage is critical for providing prognostic information, especially in the proposed G2a/G2b subdivision. Table 1WHO classifications of neuroendocrine neoplasms of endocrine organs 2025 (reference 3) and thoracic 2021 (reference 11)DifferentiationNon-thoracic organsThoracic organsTerminologyMitoses per 2mm^2^Ki67 indexTerminologyMitoses per 2mm^2^NecrosisKi67 index Well-differentiatedNET G1 < 2 < 3%TC0–1Noup to 5%NET G22 -203–20%AC2–10Focal, if anyup to 20%NET G3 > 20 > 20%Poorly differentiatedLarge cell type (LCNEC) > 20 > 20%LCNEC > 10Yes40–80%Small cell type (SCNEC) > 20 > 20%SCLC > 10Yes50–100%Mixed neoplasmsMiNENCombined SCLC / LCNEC ****TC typical carcinoid, AC atypical carcinoid, LCNEC large cell neuroendocrine carcinoma, SCLC small cell lung carcinoma, NET neuroendocrine tumor. NEC neuroendocrine carcinoma, MiNEN mixed neuroendocrine and non-neuroendocrine neoplasm. *recently divided into G2a and G2b in pancreatic NETs (ref. 25, 26, 27), ** not included in criteria, *** Each component accounts for at least 30% of total tumor cell population, **** Amount of the non-NEN components is not defined
In the 2022 WHO classification for tumors of endocrine organs, grading systems were defined or recommended for NENs of endocrine organs and non-endocrine organs [3]. Table 2 provides a synopsis of the most significant organs according to the established grading system. Additionally, it enumerates the organs for which there are recommendations regarding the assessment of proliferative activity with the exception of the thoracic organs, the established grading systems align with the grading system employed for digestive NENs. Recent studies have demonstrated the applicability of the digestive NEN grading system to pulmonary NENs [28, 29]. The recommended grading systems (see Table 2) are site-specific [3]. Efforts to establish a unified grading system for NENs across all organ systems have been initiated and are ongoing; however, as of October 2025, such harmonization has not yet been fully achieved [14]. Table 2. Grading in neuroendocrine neoplasms of different organs defined or recommended according to the 5th edition of WHO classification endocrine organs 2025OrgansGradingUse of Ki67 index and cut-off valuesUse of mitotic count and cut-off valuesGastrointestinal tractG1, G2, G3Defined (3%, 20%)Defined (2, 20 per 2mm^2^)PancreasG1, G2, G3Defined (3%, 20%)Defined (2, 20 per 2mm^2^)Thoracic organsTC (G1), AC (G2)RecommendedDefined* (2, 10 per 2mm^2^)Urogenital organsG1, G2, G3Defined (3%, 20%)Defined (2, 20 per 2mm^2^)Gynecological organsG1, G2, G3Defined (3%, 20%)Defined (2, 20 per 2mm^2^)BreastG1, G2, G3 RecommendedDefinedHead and Neck organsG1, G2, G3Defined (3%, 20%)Defined (2, 20 per 2mm^2^)Thyroidhigh grade RecommendedRecommendedParathyroidNoneRecommended**RecommendedAdrenal medullaNoneRecommendedRecommendedSkinNonenonoPituitary glandNoneRecommendedRecommended**Footnote: abbreviations: TC typical carcinoid, AC atypical carcinoid^^Presence of necrosis belongs to the grading features of thoracic NEN. ** Nottingham grading should be applied. *** High-grade medullary thyroid carcinoma is defined by ≥ 5 mitoses/2 mm^2^, Ki-67 index ≥ 5%, and/or tumor necrosis. **** Recommended for atypical adenoma and carcinoma. ***** The pheochromocytoma of the adrenal gland scaled score (PASS), the grading system for adrenal pheochromocytoma and paraganglioma (GAPP) including Ki67 and mitotic count. ****** predictive value so far unproven
Concerning the grading evolution in metastasizing NETs of the digestive or pulmonary system, several studies have demonstrated a grading increase over time and under treatment. A histological evaluation of biopsies obtained at two or more time points from patients with digestive NETs, predominantly of pancreatic and small intestinal origin, revealed an increase in the proliferation index, thereby establishing tumor progression [30–32]. A comparable observation was reported in pulmonary carcinoids, wherein 35% of Stage IV metastatic carcinoids exhibited mitotic counts exceeding 10 [33]. Subsequent studies have identified TP53 mutations in metastatic pulmonary, thymic, and pancreatic neuroendocrine tumors that have developed from G1/G2 tumors and exhibit high-grade histological transformation [34–36]. The most recent study documented that 9/40 metastasizing NETs G3 that underwent histological examination on two or more occasions during their disease exhibited in the last examination NENs with focal features consistent with partially NEC-like histology (i.e., high-grade atypia, diffuse growth pattern, and/or necrosis). An analysis of the cases revealed that, while initially classified as TP53 wild type, subsequent genetic testing identified mutations in the TP53 gene. These mutations were associated with accelerated tumor growth and a significant increase in the Ki67 index, indicating a protracted clinical course following multimodal treatment. Of the nine tumors examined, seven were of pancreatic origin and exhibited genetic characteristics indicative of PanNETs, including mutations in the MEN1 and/or DAXX gene [37]. The clinicopathological history as well as the molecular changes show that these tumors have a NET origin; consequently, these neoplasms were designated as "NEC-like G3 NET" [37]. A similar histological and genetic transformation of NETs has been also reported in the pituitary gland [38, 39].
A possible correlation between NEC-like transformations and a specific treatment regimen, e.g., peptide receptor radionucleotherapy (PRRT), alkylating chemotherapy, has been discussed [40, 41].
In the lung, it has recently been shown that 20 of 600 (3%) biopsied or resected SCLCs with wild-type RB1 and TP53 showed chromothripsis-mediated genetic alterations. In 5 of these patients with SCLCs, some tissues from the primary or nodal metastases exhibited carcinoid histology, indicating that the pulmonary high grade NENs have arisen from lower grade carcinoids/NETs showing a similar high grade transformation [42].
Subtyping of NENs
Subtyping by hormonal function
In addition to the differences between NET and NEC, also NETs are heterogeneous, a characteristic that is particularly evident in cases arising in the pancreas, stomach, duodenum, rectum, lung, and pituitary gland. Pancreatic neuroendocrine tumors (PanNETs) and pituitary neuroendocrine tumors (PitNETs) are associated with a high prevalence of hormonal syndromes (Table 3) [43, 44]. Other functioning NETs occur in the parathyroid and thyroid gland, duodenum and ileum and rarely in the stomach, jejunum and colon. In the pancreas and the pituitary gland, the separation of functioning from non-functioning NETs is still in use since they differ in clinical terms regarding therapy and outcome. PanNETs are associated with nine clinically defined hormonal syndromes (Table 3) and are named after insulinoma, glucagonoma, VIPoma etc. A few tumors such as PPoma, somatostatinoma, calcitoninoma and ghrelinoma also received terms suggesting that they are associated with a hormonal syndrome, however, these tumors cannot be clinically related to a well-defined syndrome [45, 46]. Many serotonin positive PanNETs are non-functioning and PanNETs with carcinoid syndrome appear to be extremely rare [47–49], since a closer look at the literature reveals that most of the published serotonin positive PanNETs with syndrome probably represented metastatic lesions of occult ileal NETs [50]. In the case of somatostatin positive NETs, two distinct subtypes have been identified: one located in the ampullary region and the other in the pancreas. These subtypes can be categorized as either clearly non-functioning (i.e., ampullary somatostatinoma) or as cases that are subject to debate regarding an association with a specific syndrome (i.e., pancreatic somatostatinoma) [51]. Table 3. Hormone associated syndromes in neuroendocrine tumors in the bodyHormone associated syndromeTumor typeOrganHormoneCushing syndromeACTHCorticotroph tumorPituitary glandACTH producing tumorLungACTH producing tumorPancreasACTH producing tumorThymusACTH producing tumorKidneyACTH producing tumorPheochromochytomaACTH producing tumorThyroidAcromegalySomatotroph tumorPituitary glandGHHyperprolactinaemiaLactotroph tumorPituitary glandProlactinHyperthyroidismThyrotroph tumorPituitary glandTSHHypogonadism or hypergonadismGonadotroph tumorPituitary glandLH, FSHHypoglycemiaInsulinomaPancreasInsulinGlucagonoma syndromeGlucagonomaPancreasGlucagonWDHA syndrome*VIP VIPomaPancreas VIPomaExtrapancreatic gagnglioneuromaCarcinoid syndromeSerotonin Serotonin producing tumorIleum, Jejunum Serotonin producing tumorLung Serotonin producing tumorDuodenum Serotonin producing tumorStomach Serotonin producing tumorPancreasZollinger Ellison syndromeGastrin GastrinomaDuodenum GastrinomaPancreasHypercalcemiaPTH tumorParathyroid glandPTHPTHrP producing tumorPancreasPTHrPPTHrP producing tumorLungPTHrPUlcer without hypergastrinemiaCCKomaPancreasCCKNET without clinically defined syndromePPomaPancreasPPSomatostatinomaPancreasSomatostatinSomatostatinomaAmpullaSomatostatinGhrelinomaPancreatic/extrapancreaticGhrelinMedullary thyroid carcinomaThyroid glandCalcitoninCalcitoninomaPancreasFootnote: abbreviation: ACTH adrenocorticotropic hormone, GH growth hormone/somatotrophic hormone, TSH thyroid stimulating hormone, LH luteinizing hormone, FSH follicle stimulating hormone, VIP vasoactive intestinal peptide, PTH parathormone, PTHrP parathormone-related peptide, CCK Cholecystokinin, PP pancreatic polypeptide. *watery diarrhoea, hypokalaemia, and achlorhydria
The functionality of PitNETs is associated with hypersecretion of growth hormone (GH), prolactin, thyroid-stimulating hormone (TSH), adrenocorticotropic hormone (ACTH), follicle-stimulating hormone (FSH), and luteinizing hormone (LH) [44]. Parathyroid neoplasms secrete parathormone (PTH), medullary thyroid carcinoma produces calcitonin, which may cause hypocalcemia, and functioning duodenal NETs secrete gastrin [52], and ileal NETs secrete serotonin (Table 3).
While the aforementioned hormones are orthotopic to their tumor locations, ACTH may be produced as ectopic hormone in extra-pituitary organs, most frequently in the lung [53], the pancreas [54, 55], the thymus [56] and the kidney [57].
It is noteworthy that the prevalence of functional tumors is significantly lower than that of non-functional tumors, despite the fact that non-functional tumors contain and produce hormones analogous to those of functional tumors. The distinction between these two states is attributable to the uncontrolled secretion of the produced hormone and the resulting clinical syndrome and serum level.
A special subtyping approach is applied to the NETs of the stomach. Five types of gastric histamin-producing (ECL-cell) NETs are distinguished based on various clinicopathological determinants and the tumor pathogenesis. In addition, there are NETs producing somatostatin, gastrin and serotonin (see chapter NEN types of the stomach, Annual Review Issue 2026) https://link.springer.com/article/10.1007/s00428-025-04340-x.
Subtyping by transcription factor expression
Recently, NETs have been subtyped based on transcription factor expression related to cellular hormone positivity. The organs that have profited from this way of subgrouping of NETs include pituitary gland, pancreas, lung, kidney and appendix (Table 4). Table 4. Subtyping by hormonal syndromes and differential expression of hormones and transcription factors in neuroendocrine tumors of the pituitary gland, pancreas, appendix and kidney (modified from references 57, 58, 59, 62, 63)Tumor typeHormone related symptomesHormone expressionTranscription Factor in subtypesPituitarySomatotroph tumorAcromegaly (florid or subtle)GHPIT1Lactotroph tumorHyperprolactinaemiaProlactinPIT1, ERaThyrotroph tumorHyperthyroidismTSHPIT1, GATA2/3Corticotroph tumorCushing syndromACTHTPIT, NeuroD1Gonadotroph tumorHypogonadism or hypergonadismLH, FSHSF1, ERa, GATA2/3Null cell tumourNonenoneNonePancreasNon functioningnoneVariable (frequently Glu, PP, Som)ARXInsulinomaHypoglycemiaInsulinPDX1, (ARX**)GlucagonomaHyperglucagonemiaGlucagonARX, PDX1VIPomaWDHA syndromeVIPARXGastrinomaZollinger-Ellison syndromeGastrinNDSerotonin producing tumor*Carcinoid syndromeSerotoninNDACTHomaCushing syndromeACTHPDX1, CDX2GHRHomaAcromegalyGHRHNDPTHrPomaHypercalcemiaPTHrPNDCCKomaUlcer without hypergastrinemiaCCKNDAppendixNon functioningnoneEC-cell (serotonin)CDX2, SATB2Non functioningnoneL-cell (PYY, PP, Glu)CDX2, SATB2KidneyNon functioningnoneGlucagon, PP, SomISL1, SATB2ACTH producing tumorCushing syndromeACTHnoneFootnote: abbreviation: GH growth hormone/somatotrophic hormone, TSH thyroid stimulating hormone, ACTH adrenocorticotropic hormone, LH luteinizing hormone, FSH follicle stimulating hormone, Glu Glucagon, PP pancreatic polypeptide, Som somatostatin, WDHA watery diarrhoea, hypokalaemia, achlorhydria, VIP vasoactive intestinal peptide, GHRH growth hormone releasing hormone, PTHrP parathormone-related peptide, CCK Cholecystokinin, EC-cell enterochromatin cell, PYY peptide YY, ND no data available, * unclear whether PanNET positive for serotonin are associated with a carcinoid syndrome. ** in metastatic insulinomas
In pituitary NETs, three distinct transcription factors have been identified: PIT1 (pituitary-specific positive transcription factor 1), TPIT (T-box transcription factor), and SF1 (steroidogenic factor 1) are critical in defining PitNETs in conjunction with GATA3, estrogen receptor alpha (ERa), and hormones. This complex interplay results in the classification of six adenohypophysial cell types, each of which exhibits a multitude of morphological subtypes, thus creating a comprehensive and nuanced understanding of the diverse tumor types present [3, 58] (Table 4).
In pancreatic NETs, transcription factor expression patterns, especially ARX (aristaless related homeobox) and PDX1 (pancreatic and duodenal homebox 1), reflect islet cell lineages. PanNETs with ARX expression shows frequently MEN1/DAXX/ATRX mutations and methylation patterns with similarity to alpha cells from normal pancreatic islets, while PanNETs with PDX1 expression and typically insulinomas show DNA methylation patterns with similarity to normal beta cells. Beta cell like PanNETs show usually no MEN1/DAXX/ATRX mutations [59–61] (see chapter “Novel Concepts of cell-of origin in NEN”, Annual Review Issue 2026) https://link.springer.com/article/10.1007/s00428-025-04311-2. Together with ARX and PDX1, ISL1 (insulin gene enhancer protein), CDX2 (homeobox protein CDX2), either alone or in combination with each other define five subgroups (subgroup A1, A2, B, C, D in Fig. 2). Two of these subgroups (mostly positive for ARX and ISL1, subgroups A1 and A2 in Fig. 2) resemble the previously by genetic and epigenetic studies described alpha cell like group [59] and strongly relate to glucagon and PP expression and trabecular-reticulated histological pattern, while the other subgroup (subgroup B with PDX1 and ISL1 expression in Fig. 2) resembles the previously described beta cell like type [59, 61] and expresses insulin and somatostatin [62]. The tumors of this group show predominantly solid pattern and include all insulinomas. The remaining two subgroups either express none of the four transcription factors (subgroup C in Fig. 2) or PDX1 in addition to CDX2 (subgroup D in Fig. 2). The tumors of these subgroups mainly express hormones such as serotonin, calcitonin and ACTH and have a solid pattern [62] (Fig. 2, Table 4). In pulmonary NETs, the transcription factors ASCL1 (achaete-scute homolog 1), OTP (orthopedia homebox) and HNF1A (hepatocyte nuclear factor 1) identify three major pulmonary carcinoid subgroups that show distinct clinical features such as sex, age and location [63]. In another study, the expression profiles of the transcription factors OTP and ASCL1 allow classification into four distinct groups that differ in histology (solid, trabecular, spindle cell, oncocytic), hormone expression (ACTH, GRP [gastrin-releasing peptide], serotonin, calcitonin), and clinical outcome. OTP+/ASCL1+ tumors typically exhibit solid and spindle-cell morphology with diffuse GRP and frequent ACTH expression. ASCL1-negative tumors are characterized by a trabecular growth pattern, whereas OTP-negative tumors occasionally display oncocytic morphology. These findings underscore the key role of OTP and ASCL1 in defining the molecular and morphological heterogeneity of pulmonary NETs [64].Fig. 2. Subtypes of pancreatic neuroendocrine tumors based on expression signatures of four transcription factors, ISL1, ARX, PDX1 and CDX2, and their characteristic histological, hormonal and molecular features and outcome of the patiants. Abbreviations: TF transcription factor, PP pancreatic polypeptide. Footnote: results based on the reference 63. *A2 expressing ARX, ISL1 and partly PDX1. ** including 17 insulinomas
In appendiceal NETs, differential immunostaining for hormones distinguished between the frequent serotonin positive (EC-cell type) type and the rare PYY/glucagon positive (L-cell type) type, both types, however, expressed the transcription factors CDX2 and SATB2 (Table 4) [65] (Table 4).
In renal NETs, transcription factor subtyping revealed that non-functioning NETs were comprised of glucagon, somatostatin, and PP cells, which expressed the transcription factors ISL1 and SATB2. In contrast, functioning NETs (all with ectopic Cushing syndrome) were negative for ISL1, SATB2, PAX8, and CDX2, and expressed ACTH [57].
However, a paucity of comprehensive data exists regarding the correlation between hormone phenotype and transcription factor expression in certain studies of rectal [66] and duodenal NETs [67]. In the duodenum, ampullary regions, and rectum, hormone-defined subtypes have been described; however, these subtypes have not been correlated with differential transcription factor expression [68–70].
Recent studies have identified subtypes of SCLC based on the differential expression of four transcription factors: ASCL1, NeuroD1, YAP1 and POU2F3. The most prevalent subtype is identified as SCLC type A, characterised by predominant ASCL1 expression, accounting for 40–50% of all SCLCs. The second most common subtype is SCLC type D, which is distinguished by the expression of NeuroD1, accounting for 20–30% of cases. These subtypes exhibit a high neuroendocrine marker expression. SCLC- P (POU2F3 expression) and SCLC-Y (YAP1 expression) are both extremely rare, accounting for 5–15% of cases respectively and express only weak neuroendocrine marker expression [71–73]. In these studies, neuroendocrine differentiation was assessed by immunohistochemistry for neuroendocrine markers (e.g., chromograninA, synaptophysin) [73]; in some studies corroborated by RNA based neuroendocrine signature scores [74].
In the context of LCNEC of the lung, the recognition of two distinct subtypes has been made, with this categorisation based on the profile of specific transcription factors as well as genetic alterations. About half of LCNEC exhibits elevated levels of ASCL1 and DLL3 expression, biallelic mutations in TP53 and RB1, and pronounced neuroendocrine features. It has been demonstrated that the molecular features of these LCNEC exhibit significant overlap with those of SCLC and therefore is designated as SCLC-like LCNEC. The remaining LCNEC exhibits low expression of ASCL1 and DLL3, frequent TP53 and STK11/KEAP1 mutations, and incomplete or weak neuroendocrine differentiation, and is designated as NSCLC-like SCLC [19, 75].
In the field of extrapulmonary NECs, transcription factor-based subgrouping has been explored [76, 77], including NECs of the digestive organs [77], genitourinary or gynaecological organs [77–80] and olfactory neuroblastomas [81]. The relationship between these subtypes and the treatment management of pulmonary and extrapulmonary NEC patients is still debated.
Subtyping of NETs by genetic alterations
The subtyping of NETs based on genetic alterations and profiles highlights only few gene mutations, either in the sporadic setting or in the hereditary diseases. The most frequently sporadically mutated gene is MEN1, which is found in decreasing frequency in the endocrine pancreas (35–40%), lung (11–18%), thymus (5–10%), parathyroid and duodenum (rare).
In the pancreas, MEN1 cooperates frequently with DAXX/ATRX (approx. 35%). DAXX/ATRX mutations have been observed to promote alternative lengthening of telomeres (ALT) and chromosomal instability (CIS) [82, 83]. Presence or absence of MEN1/DAXX/ATRX mutations of PanNETs are closely linked to their methylation patterns similar to alpha- and beta-cells [59–61] (see above and see chapter “Novel Concepts of cell-of origin in NEN”, Annual Review Issue 2026) https://link.springer.com/article/10.1007/s00428-025-04311-2. Mutations of DAXX/ATRX, ALT and/or CIS are related to poor outcome of the PanNET patients [61, 83–86]. Integrative analyses of transcriptomic and proteomic studies have identified subtypes of PanNETs that partially align with alpha- and/or beta-cell-like signatures [87–89]. mTOR pathway genes (e.g., TSC2, PTEN, PIK3CA) may play a pathogenetic role in approx. 15% of sporadic PanNETs and VHL may be pathogenetically involved in only a few percent [90]. In 30% of insulinomas, gene YY1 has been found to be mutated and seems to be independent from MEN1 gene mutations of pathogenetic significance [91]. Metastatic insulinomas, which account for 10% of all insulinomas, are distinguished from non-metastatic insulinomas by high ALT and expression of ARX [92].
In pulmonary NETs, the MEN1 gene mutations are rare (11–18%) and typically occur in the absence of DAXX/ATRX mutations [20, 21, 61, 90, 93–95]. Atypical carcinoids (ACs) are more frequently affected than typical carcinoids (TCs) [20, 93, 95]. Several other genes (e.g., chromatin remodeling genes, mTOR pathway genes) have been reported but as their recurrence rate is infrequent, their pathogenetic role is unclear [20].
In the stomach and rectum, where sporadic MEN1 mutations are extremely rare or even absent, there are no data that identified genes with pathogenetic significance. There are no data of gene mutations that could qualify as driver genes [96–98]. There are no sufficient data on genetic features of duodenal and colonic NET.
In PitNET, sporadic MEN1 mutations seem to be extremely rare. The most frequent altered gene is GNAS, which is affected in 40–60% of somatotroph tumors, followed by USP8 gene, which is found in 20–60% of corticotroph PitNETs [99, 100].
A distinctive genetic configuration is observed in medullary thyroid carcinoma, which, in sporadic cases, is marked by either a RET mutation at codon M918T or an RAS (HRAS, KRAS, NRAS) mutation with RET mutation, indicating a poor prognosis, while RAS is associated with a favourable prognosis [101].
The genetic landscape of the jejunoileal NETs is unique and distinct from those of all other NENs of the body. Somatic mutations are rare and mainly concern the CDKN1B gene. Other driver mutations are missing [102] However, chromosome 18 deletions are frequently observed in 60–90% of the cases, along with epigenetic changes [102]. Subtyping the molecular profile of jejunoileal NETs revealed three prognostically distinct subgroups [102]. A favourable prognosis was associated with chromosome 18 loss, CpG islet methylator phenotype negativity, and CDKN1B mutation. CpG islet methylator phenotype positivity and the absence of whole-arm copy-number variation were associated with intermediate prognosis, while the presence of multiple whole-arm copy-number variation were associated with poor prognosis. The distinctive configuration of the jejunoileal NETs is accentuated by the near absence of jejunoileal NECs, a characteristic that is also observed in appendiceal NENs [43].
Subtyping of NECs by genetic alterations
As already discussed in “dichotomy of NENs”, NECs are distinct from NETs by their common association with TP53 and/or RB1 mutations. In addition, pulmonary NECs are distinguished by high tumor mutation burden (TMB) and copy number variation (CNV) [76], which is less frequent in GEP-NEC [103]. Furthermore, the digestive NECs, and also mixed neuroendocrine-nonneuroendocrine neoplasms (MiNENs), harbor mutations in genes such as KRAS and BRAF, with frequencies that exhibit significant variations depending on the specific location of the tumor (see Table 5). Up to 50% of GEP-NEC harbour potentially targetable molecular alteration, including BRAF mutations and mismatch repair (MMR) protein deficiency associated with microsatellite instability (MSI) in colorectal NECs [103, 104]. Many of the reported molecular changes in GEP-NECs also apply to MiNEN/MANECs [76]. In the lung, a part of LCNECs and a very small population of SCLC share genetic features with NSCLC, e.g. adenocarcinoma or squamous cell carcinomas [105]. Table 5. Common molecular alterations in NEC/MiNENsfacross different organ sitesGenes/OrgansLungStomachColorectumPancreasSCLCLCNECTP53 mutation90–95%90–95% in SCLC-like, 35–50% in NSCLC-like60–90%70%70–90%RB1 mutation/deletion90–95%30–50%40–60%60–90%KRAS mutationvery rare~20%~15%30–60%50–90%BRAF mutationvery rarevery rarevery rare10–20%very rareAPC mutationvery rarevery rarevery rare30–50%very rareARID1A mutationrarevery rare40-50%10–20%10–20%SMAD4 mutationvery rarerarevery rarevery rare10–40%MYC family amplification10–30%5–10%Substantial5–10%10–20%mTOR/AKT/PI3K pathway genes5–10%5–10%5–10%5–15%10–20%STK11/KEAP mutation or deletionrare20–35%**NDNDvery rareMSI-H/dMMRvery rarevery rarevery rarerarevery rareTMBhighhighmoderatemoderatelow to moderateDLL3 high expression70–85%40–50%** variablevariablevariableTagetable alterationsin clinical useDLL3, CDK4/6**NSCLC-like LCNEC: off-label use of NSCLC-targeted therapies (EGFR, ALK, BRAF, PD-L1, KRAS G12C) ************in trials/experimentalDLL3, B7-H3, TROP2, VEGFR/FGFR, Fucosyl-GM1DLL3, PD-L1, PARPsPD1, PD-L1, VEGFR, HER2PD1, PD-L1, VEGFR, MGMT, HER2, RETPD1, PD-L1, MSI-H, PARP, VEGFR/PDGFR/FGFRFootnote: abbreviation: NEC neuroendocrine carcinoma, MiNEN mixed neuroendocrine and non-neuroendocrine neoplasms, SCLC small cell lung carcinoma, LCNEC large cell neuroendocrine carcinoma, ND no data or no sufficient data available, MSI-H high microsatellite instability, dMMR mismatch repair deficiencty, TMB tumor mutation burden, ICI immune checkpoint inhibitor: Very rare < 5%, rare 5–10%, * potentially therapeutic targetable molecule, ** enriched in NSCLC-like subtype of LCNEC, ***LCNEC is classified under NSCLC for therapeutic purposes; however, targeted agents are approved for NSCLC, not specifically for LCNEC, ****no therapies specifically approved for NEC
Mixed NENs
The heterogeneity of NENs is underpinned by the occurrence of mixed neoplasms. These neoplasms have in addition to a neuroendocrine component, other non-neuroendocrine component, such as adenocarcinoma or squamous cell carcinoma. Accordingly, the tumors are named mixed neuroendocrine-nonneuroendocrine neoplasms (MiNENs). They are common in the stomach, colon, lung, prostate, and probably also in the uterus and urinary bladder, but rare in the pancreas as well as in the head and neck region. The MiNENs are discussed in detail in chapter “How to diagnose MiNEN and what are amphicrine carcinomas”, Annual Review Issue 2026 https://link.springer.com/article/10.1007/s00428-025-04241-z. Within the digestive system, the components of the MiNENs are arbitrarily quantified, with each component being required to account for more than 30% of the tumor tissue. In all other organs, no quantitation of the components is mandatory.
Hereditary diseases
Hereditary tumor diseases are relatively common in NENs and are usually associated with NETs and not with NECs. Table 6 provides a synopsis of the most salient hereditary diseases with regard to their particular genetic and morphological manifestations. Approximately 5–10% of all NETs occur in the setting of a hereditary syndrome, including MEN1, MEN2, VHL, NF1, and TSC (tuberous sclerosis). Hereditary NETs often present at a younger age, higher rates of multifocality, and a more indolent course compared to sporadic NETs [106]. Table 6. Common NET related hereditary diseases and their manifestations (modified from reference 2)Disease/phenotypeGene(s)Encoded proteinMost common manifestationsMultiple endocrine neoplasia type 1MEN1MeninParathyroid neoplasm, PitNET, DuoNET, PanNET, Thoracic NETMultiple endocrine neoplasia type 2A (formerly called type 3)RET (codon 634)RETMTC, PC, parathyroid neoplasmMultiple endocrine neoplasia type 2BRET M918TRETMTC, PC, ganglioneuromas oral/intestinal, marfanoid body habitusMultiple endocrine neoplasia type 4CDKN1Bp27Parathyroid neoplasm, PitNET, GEP NETMultiple endocrine neoplasia type 5MAXMAXPPGL, PitNETs, parathyroid neoplasm, ganglioneuroma, neuroblastomaVon Hippel–Lindau syndromeVHLVHLCNS haemangioblastoma, renal cyst, RCC, PPGL, PanNETs, SCN, DuoNET, epididymal papillary cystadenomasSDH-deficient tumour syndromes (PGL1-5)SDHD, AF2, C, B, ASDHPPGL, GIST, RCC, PitNETNeurofibromatosis type 1NF1Neurofibromincafé-au-lait spots, cutaneous neurofibromas, MPNSTs, PC, AmpNETCarney complexPRKAR1APRKAR1APPNAD, PitNET, thyroid cyst and carcinoma, Lentiginosis, Blue naevi, Myxomatosis (cardiac and cutaneous), Large cell calcifying Sertoli cell tumoursGlucagon cell hyperplasia and neoplasia (Mahvash disease)GCGRGCGRMultifocal glucagon positive micro and macro PanNETFamilial hyperinsulinismMAFA*MAFAMultifocal insulinomasFootnote: Abbreviation: NET neuroendocrine tumor, PitNET pituitary NET, DuoNET duodenal NET, PanNET pancreatic NET, MTC medullary thyroid carcinoma, PC pheochromocytoma, GEP NET gastroenteropancreatic NET, PGL1–5, paraganglioma syndrome types 1–5; SDH, succinate dehydrogenase; VHL, von Hippel–Lindau syndrome, PPGL paraganglioma phaeochromocytoma, CNS central nerve system, RCC renal cell carcinoma, SCN serous cystic neoplasms of the pancreas, MPNST, malignant peripheral nerve sheeth tumor, AmpNET ampullary NET, PPNAD primary pigmented nodular adrenocortical disease. * the very rare hereditary disorders such as Hyperparathyroidism–jaw tumour syndrome, McCune–Albright syndrome, DICER1 syndrome, are not shown in this table
There is a considerable variability in penetrance and age of first presentation among the various manifestations of hereditary diseases. For instance, in cases of MEN1, the most prevalent and earliest manifestation is hyperparathyroidism due to parathyroid adenoma, with a lifetime penetrance of over 90%. This is followed by PanNETs (60–80%) and pituitary adenoma (30–50%) [107]. In cases of VHL disease, non-neuroendocrine diseases, such as retinal or cerebral hemangioblastomas and renal cell carcinomas are most frequently observed in the age group of 20–40 years, while PanNETs are rather rare, with a penetrance rate of 10–20% and median age of 34 years [108], the occurrence of NET in VHL depends on specific type of VHL-mutations leading to HIF2 accumulation (VHL Type 2).
The reasons for underlying factors contributing to the variable penetrance and age manifestation of hereditary diseases remain unknown. There is also no explanation for the site-specificity of the lesions and the specific endocrine cell types involved [109]. In contrary, regarding tumorigenesis and tumor development the hereditary diseases provide insights into the developments of the tumors by the possibility to study the precursor lesions and their pathogenesis, as it was shown in the pancreas and the duodenum of MEN1 patients [110, 111]. Precursor lesions can be also identified associated with medullary thyroid carcinoma [101]. Glucagon cell hyperplasia and neoplasia (GCHN), otherwise known as Mahvash disease, is a condition characterized by the loss of function mutation in the glucagon receptor gene (GCGR). This hereditary mutation results in a hyperplasia-neoplasia sequence with proliferation of non-neoplastic glucagon cells within the islets and subsequent development of neoplastic glucagon cells. The somatic acquisition of a MEN1 mutation within the hyperplastic glucagon cells is believed to be a pivotal step in the neoplastic progression of this disease [112–114]. In familial insulinomatosis, an additional recently identified hereditary disease, the causative mutation impacts the MAFA gene, which is specific to the beta cell. This results in dysplastic beta cell cords that subsequently transform into insulinomas [115].
Recent studies in digestive and/or pulmonary NENs documented a multitude of germline mutations and germline pathogenic variants in addition to the classical endocrine tumor genes. These include MUTYH, CDKN1B, CHEK2, BRCA2, MYOC, ERCC2, and ERCC3, among others [21, 116–118]. The findings appear to be not specific for NETs but were also identified in colorectal or pulmonary NECs [116, 119]. In two families with small intestinal NETs extended genetic examination revealed germline MUTYH mutations but a clear family history could not be assigned to the patients with newly identified germline mutations [120].
Conclusions
Despite their profound heterogeneity, NENs adhere to a unifying concept that is evident in most anatomical locations, albeit with varying degrees of clarity. The concept delineates two distinct families of neoplasms, namely NETs and NECs, distinguished by their unique morphologies, biological characteristics, genetic profiles and prognoses.
In order to address the challenges posed by intertumoral heterogeneity in NETs, a grading system was developed. The efficacy of this system in distinguishing between NETs has been well-documented, facilitating the determination of patient outcomes and the optimal therapeutic management. Subsequent refinement of the NENs resulted in the emergence of the subtypes of NETs, which were distinguished by their functionality, genetic, epigenetic, and proteomic profiles, as well as the expression of transcription factors. These efforts have led to significant advancements in subtyping and therapeutic management, contributing to a more comprehensive understanding of the disease.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Board. W Co TE (2025) Endocrine and neuroendocrine tumours [Internet]. Lyon (France): International Agency for Research on Cancer; 2025 (WHO classification of tumours series, 5th ed.; vol. 10). Available from: https://tumourclassification.iarc.who.int/chapters/53.
- 2Travis WD, Brambilla C, Burke AP, Marx A, Nicholson AG (2015) WHO classification of tumours of the lung, pleura, thymus and heart. International Agency for Research on Cancer 10.1097/JTO.000000000000066326291007 · doi ↗ · pubmed ↗
- 3Eren OC, Bagci P, Balci S, Ohike N, Saka B, Sokmensuer C, Leblebici CB, Xue Y, Reid MD, Krasinskas AM, Kooby D, Maithel SK, Sarmiento J, Cheng JD, Taskin OC, Kapran Y, Tarcan ZC, Luchini C, Scarpa A, Basturk O, Adsay NV (2024) Subgrading of G 2 Pancreatic Neuroendocrine Tumors as 2A (Ki 67 3% to < 10%) Versus 2B (10% to </= 20%) Identifies Behaviorally Distinct Subsets in Keeping with the Evolving Management Protocols Annals of surgical oncology. 10.1245/s 10434-024-15632-y 10.1245/s 10434-024-15632- · doi ↗ · pubmed ↗
- 4Kasajima A, Konukiewitz B, Oka N, Suzuki H, Sakurada A, Okada Y, Kameya T, Ishikawa Y, Sasano H, Weichert W, Kloppel G (2019) Clinicopathological Profiling of Lung Carcinoids with a Ki 67 Index > 20 Neuroendocrinology 108:109–120. 10.1159/00049580610.1159/00049580630485860 · doi ↗ · pubmed ↗
- 5Merola E, Perren A, Rinke A, Zerbi A, Mc Namara MG, Arsenic R, Fazio N, de Herder W, Valle JW, Gress TM, Wiedenmann B, Pascher A, Pavel ME (2022) High rate of Ki-67 increase in entero-pancreatic NET relapses after surgery with curative intent J Neuroendocrinol 34:e 13193. 10.1111/jne.1319310.1111/jne.1319336306194 · doi ↗ · pubmed ↗
- 6Cordero-Hernandez IS, Ross AC, Dasari A, Halperin DM, Chasen B, Yao JC (2024) Transformation of G 1-G 2 neuroendocrine tumors to neuroendocrine carcinomas following peptide receptor radionuclide therapy Endocr Relat Cancer 31. 10.1530/ERC-23-020310.1530/ERC-23-020338329269 · doi ↗ · pubmed ↗
- 7Leunissen DJG, Moonen L, von der Thusen JH, den Bakker MA, Hillen LM, van Weert TJJ, Zur Hausen A, van den Bosch TPP, Lap LMV, Damhuis RA, Reynaert NL, van den Broek EC, group P, Fernandez-Cuesta L, Foll M, Alcala N, Sexton-Oates A, Dingemans AC, Speel EM, Derks JL (2024) Identification of Defined Molecular Subgroups on the Basis of Immunohistochemical Analyses and Potential Therapeutic Vulnerabilities of Pulmonary Carcinoids J Thorac Oncol. 10.1016/j.jtho.2024.11.01810.1016/j.jtho.2024.11.01839 · doi ↗ · pubmed ↗
- 8Ura A, Evert K, Evert M, Märkl B, Kremer M, Moser E, Sasano H, Okada Y, Steiger K, Mogler C, Jesinghaus M, von Werder A, Safi S, Hoffmann H, Klöppel G, Kasajima A (2025) Phenotypic landscape of pulmonary neuroendocrine tumors: subtyped by otp/ascl 1 expression correlated with histology, hormones and outcome. Endocr Pathol 36:43. 10.1007/s 12022-025-09882-z 10.1007/s 12022-025-09882-z PMC 1259224641196447 · doi ↗ · pubmed ↗