Diagnosis and grading of adrenal cortical carcinoma
Giulia Vocino Trucco, Eleonora Duregon, Mauro Papotti, Marco Volante

TL;DR
This paper reviews the updated WHO classification for adrenal cortical carcinoma, combining histopathology and molecular insights to improve diagnosis and grading.
Contribution
The paper provides a practical diagnostic framework integrating molecular and histopathological criteria for adrenal cortical carcinoma.
Findings
The WHO classification uses multiparametric scoring to assess invasion, architecture, and mitotic activity for ACC diagnosis.
A two-tiered grading system based on mitotic count improves consistency in adrenal cortical carcinoma evaluation.
Immunohistochemistry with markers like SF1 and Ki-67 aids in confirming diagnosis and predicting outcomes.
Abstract
The 5th edition of the WHO classification of endocrine and neuroendocrine tumors represents a significant advancement in the diagnostic approach to adrenocortical carcinoma (ACC), integrating novel molecular insights with established histopathological criteria to enhance diagnostic accuracy and to refine prognostic assessment. This review outlines key histopathological features and diagnostic strategies for ACC, offering a practical framework for evaluation and grading in daily practice. The updated WHO classification reaffirms the central role of histopathology, employing multiparametric scoring systems that assess invasion, architectural and cytological features, mitotic activity, and necrosis. However, these parameters often pose interpretive challenges, and no single algorithm ensures complete sensitivity, specificity, or reproducibility. Therefore, combining diagnostic approaches…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
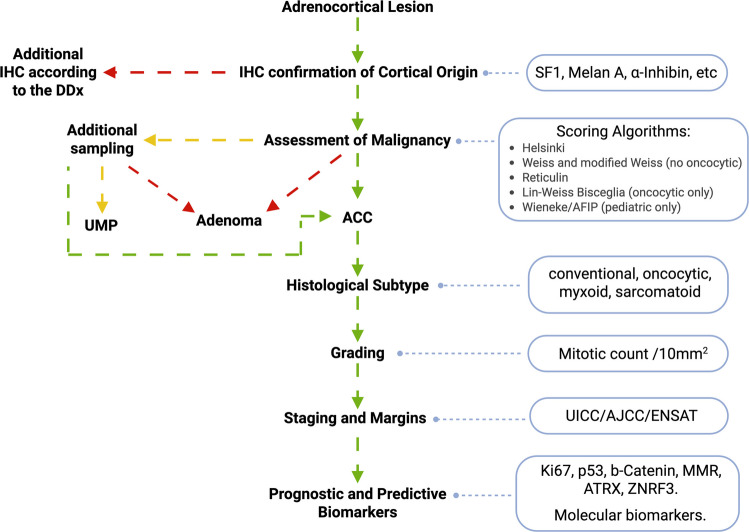 Figure 1
Figure 1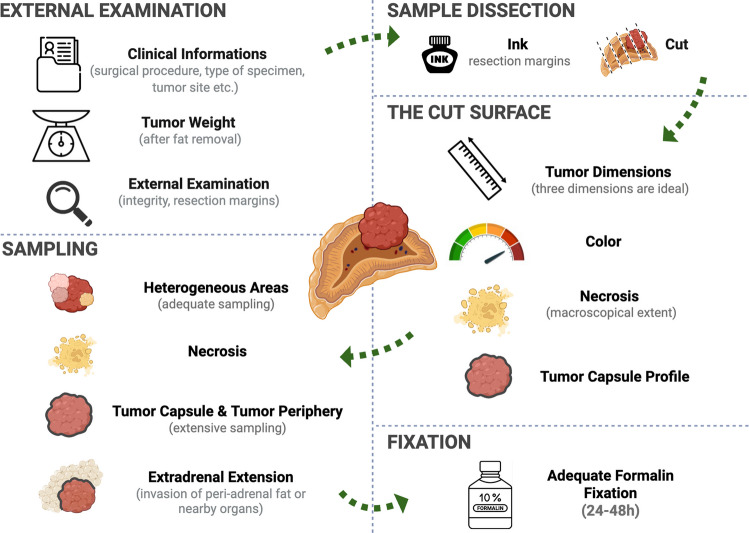 Figure 2
Figure 2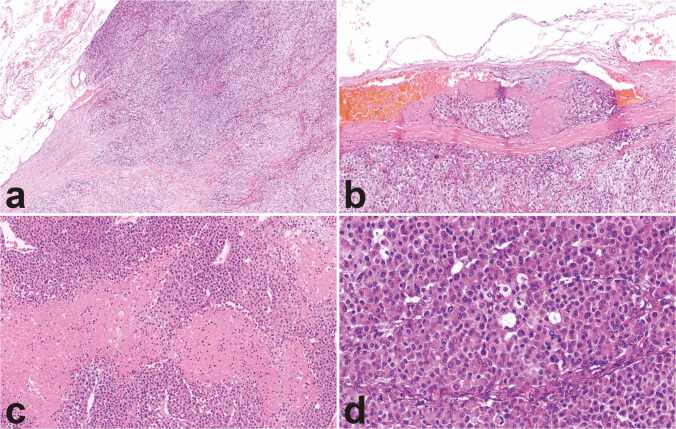 Figure 3
Figure 3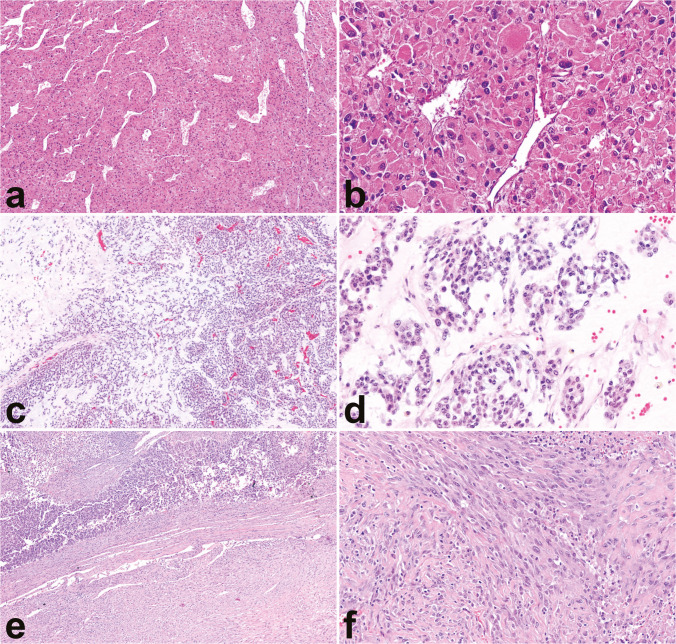 Figure 4
Figure 4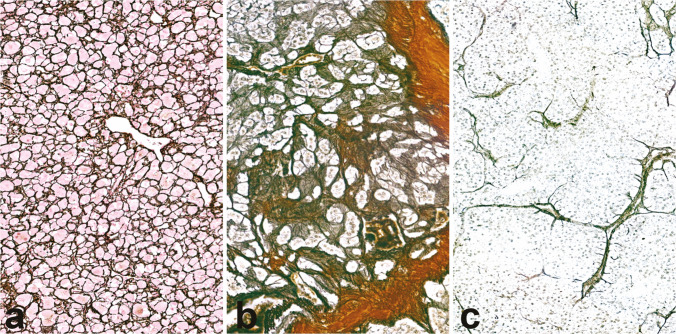 Figure 5
Figure 5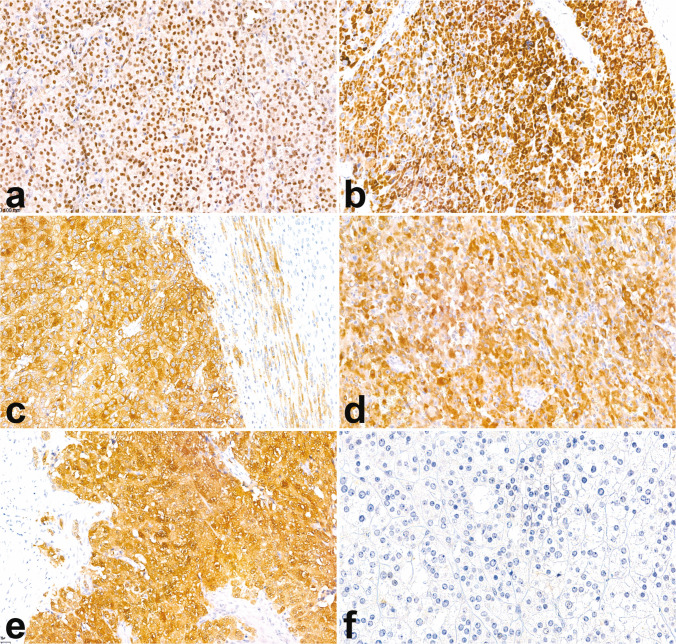 Figure 6
Figure 6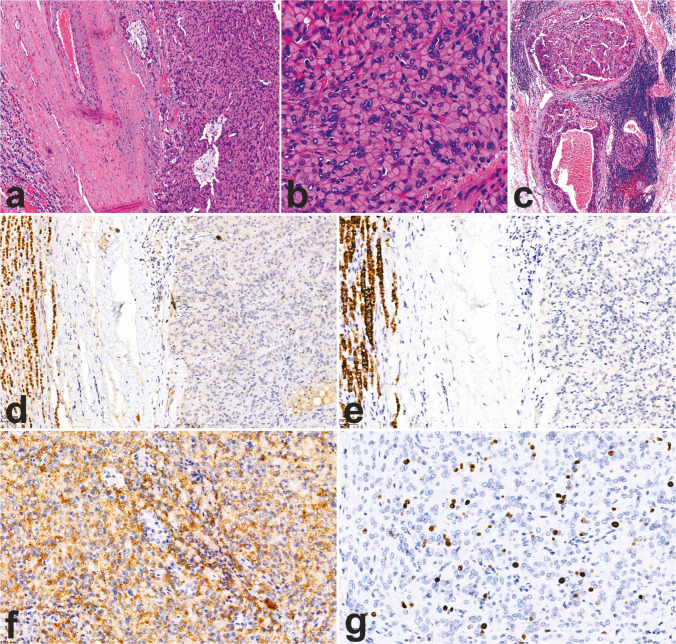 Figure 7
Figure 7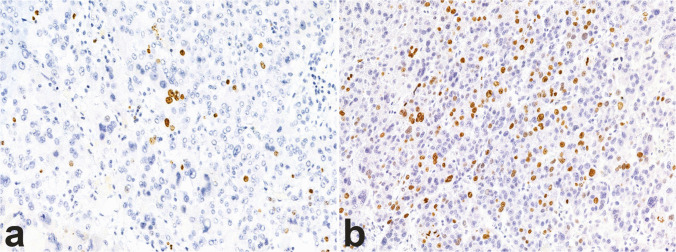 Figure 8
Figure 8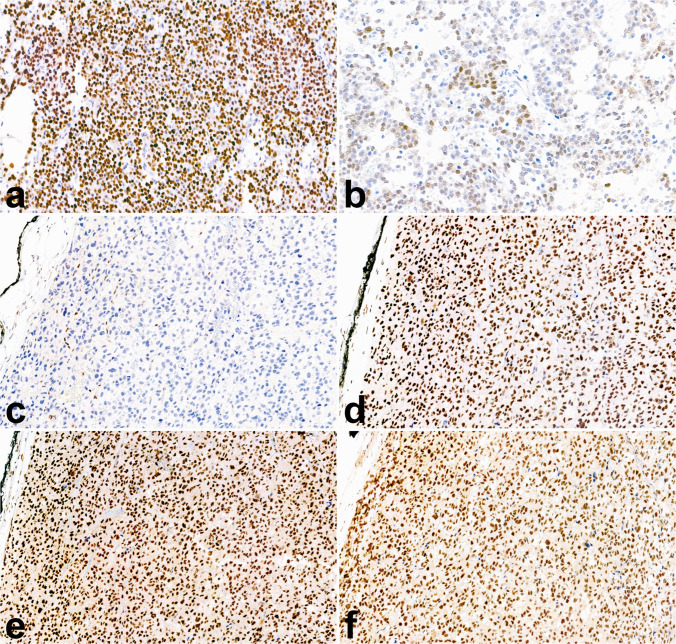 Figure 9
Figure 9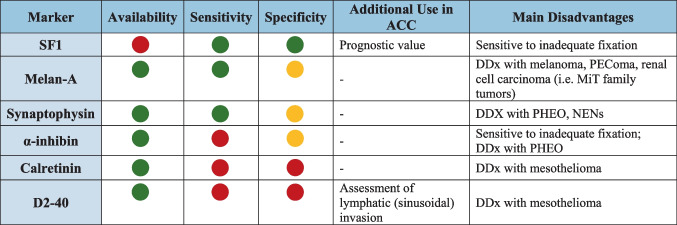 Figure 10
Figure 10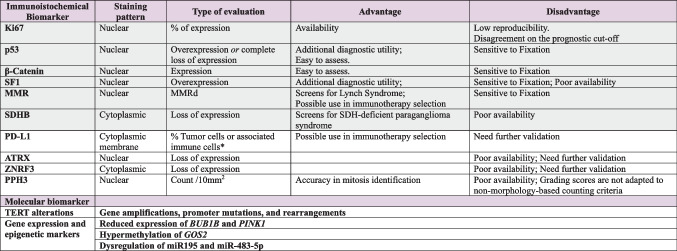 Figure 11
Figure 11- —http://dx.doi.org/10.13039/501100005010Associazione Italiana per la Ricerca sul Cancro
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAdrenal and Paraganglionic Tumors · Cancer, Hypoxia, and Metabolism · Pituitary Gland Disorders and Treatments
Adrenal cortical carcinoma: a brief introduction
Adrenal cortical carcinoma (ACC) is a malignant tumor arising from adrenal cortical cells. It is a rare disease with an estimated incidence of 1 case per million in adults and 0.3 cases per million in children [1]. ACC is an aggressive disease with a dismal prognosis, accounting for the majority of deaths attributable to primary adrenal neoplasia [2] and with an estimated 5-year overall survival rate between 37 and 47% [3, 4].
ACC is usually a unilateral disease, with a preferential localization in the left adrenal gland [1, 5], whereas synchronous or metachronous bilateral involvement is rare. Exceptionally, ACCs may also occur in ectopic locations, and hitherto sporadic cases have been reported in the retroperitoneum, pelvic region, and ovary [6, 7].
Most ACC cases in adults occur sporadically. In this context, the most relevant etiologic factor is tobacco smoking [8, 9] with a two-fold greater incidence in smokers, which is even more pronounced in males.
Importantly, ACC is associated with a significant history of previous or subsequent associated cancers, thus suggesting heterogeneous underlying cancer predisposition mechanisms [9]. Associated malignancies are extremely variable, including different types of carcinomas, as well as testicular germ cell tumors, melanomas, lymphomas, and sarcomas. A proportion of ACC cases occur in the context of several germline susceptibility syndromes [10, 11]. Most commonly, these syndromic ACC cases were found in Li–Fraumeni syndrome, accounting for 3–5% of adult ACC cases [10, 11] and 50–80% of pediatric cases [10], but also Lynch syndrome [12, 13], Carney complex [14], familial adenomatous polyposis (FAP) [15], Beckwith–Wiedemann syndrome [16], multiple endocrine neoplasia type 1 (MEN1) [17], neurofibromatosis type 1 [18], and possibly subsets of the familial paraganglioma phaeochromocytoma syndromes [19], FH (hereditary leiomyomatosis and renal cell carcinoma syndrome) [20] or synchronous MSH2 and RET variants (without multiple endocrine neoplasia type 2) [21].
Most patients seek medical attention for symptoms related to hormone hypersecretion or for symptoms secondary to the compressive effects of an abdominal mass [22, 23]. However, with the increased adoption of advanced imaging techniques worldwide, a growing number of ACCs are now incidental findings and currently account for about 10% of cases [22].
The WHO Classification (5th edition): a structural update
The 5th edition of the World Health Organization (WHO) classification of endocrine and neuroendocrine tumors introduced revisions to the diagnostic framework for ACC, integrating molecular insights with histopathological criteria to enhance diagnostic precision and prognostic relevance. These revisions aligned histological evaluation with contemporary molecular advancements in the fields of endocrine pathology, oncology, and molecular biology, offering a conceptual framework for tailored risk assessment and personalized management of ACC.
Importantly, the histopathological features remain the cornerstone of the diagnosis, and the main pathological characteristics as well as diagnostic tools are described in detail in this review.
Additionally, the WHO classification 5th edition emphasizes the importance of accurate proliferation metrics, such as mitotic counts and Ki-67 index. A significant update in the current classification is the shift from reporting mitotic count per high-power fields (HPFs) to a standardized area measured in mm^2^ addressing the well-known variability in field size across different microscopes from areas with highest mitotic density, even if these are found on different slides [2]. Lastly, the 5th edition of WHO classification emphasizes the role of diagnostic and predictive immunohistochemical biomarkers that will be discussed in detail in the present review. Additionally, it expands to transcriptome [24] and pangenomic analyses [25], as well as methylation profiling which may provide prognostic information [26], highlighting their emerging role in the molecular risk stratification of ACCs [27–29].
Diagnostic approaches in ACC
A practical diagnostic algorithm is shown in Fig. 1.Fig. 1. Diagnostic algorithm for the microscopical assessment of ACC. The arrows are color-coded: green refers to positive results, yellow to doubtful results, and red to negative results. IHC, immunohistochemistry; DDx, differential diagnosis; UMP, uncertain malignant potential; ACC, adrenal cortical carcinoma
General macroscopy
ACC often presents as a large solitary adrenal mass, with a mean size of about 11 cm (range 1.6 to 30 cm) [5] and weighs around 350 g (range 4 to 3500 g) [30, 31].
Most ACCs are surrounded by a prominent fibrous capsule, and the macroscopic assessment of capsular integrity is important for diagnostic and prognostic purposes [32]. The demonstration of capsular invasion is also a key criterion in several multifactorial diagnostic scoring systems [33–35]. In addition to the identification of capsular invasion, generous sampling of the tumor capsule also allows for an adequate assessment of other types of invasion such as vascular and lymphatic “sinusoidal” invasion [32], which must be evaluated at or beyond the capsular edge [2, 32]. It is important to keep in mind that the probability of finding invasive foci is largely dependent on the extent of sampling; therefore, adequate tissue sampling should prioritize the tumor periphery and capsule over central tumor areas [32]. If evident, tumor extension beyond the tumor capsule into peri-adrenal soft tissues, large veins, or nearby organs should be promptly documented, as these aspects define stage III and stage IV disease [36]. Lastly, inking the surgical specimen before sectioning and careful assessment of tumor margins should always be performed.
On the cut surface, ACC usually presents as a vaguely nodular, yellowish-tan mass, eventually interspersed with areas of hemorrhage and necrosis. Nonetheless, a certain degree of heterogeneity is frequently observed, as a direct reflection of the diverse cellular composition and histological patterns intermingling within a single lesion. Therefore, accurate and exhaustive sampling of the tumor is recommended in order for it to be representative of its wide tumor heterogeneity.
An overview of the macroscopic assessment process is summarized in Fig. 2.Fig. 2. Overview of the macroscopic assessment process
General histopathology
Capsular invasion, despite its importance, has currently no universally accepted definition. Some authors regard any breach of the capsule as indicative of invasion, while others require full-thickness penetration to meet this criterion [32]. Moreover, its identification can be challenging due to irregularities in the capsule and the presence of fibrous interconnecting septa extending into the tumor. In contrast, direct invasion into peri-adrenal fat or adjacent organs constitutes definitive evidence of malignancy, as it reflects tumor extension beyond the adrenal capsule (Fig. 3a). In cases where the tumor completely breaches the capsule, it remains unclear whether a stromal reaction within the periadrenal fat is necessary to confirm periadrenal fat invasion, or if the mere presence of tumor cells within the periadrenal fat (with absent stromal response) should be considered sufficient evidence of such invasion.Fig. 3. Microscopic features of ACC. Microscopic features suggestive of malignancy include capsular invasion and extracapsular extension (a, 10×), as well as angioinvasion, defined by tumor cells associated with fibrin thrombi in capsular (b, 20×) or extracapsular vessels. Other features associated with malignancy are coagulative necrosis (c, 25×), elevated mitotic count, and atypical mitoses (bottom left, a tripolar mitotic figure) (d, 40×)
Vascular invasion is assessed at the intersection of the tumor and adrenal capsule or beyond the adrenal capsule and should be distinguished in angioinvasion and sinusoidal invasion [2]. Even though gross or clinically apparent large vessel involvement has become an uncommon finding [32], data guiding the assessment of microscopic angioinvasion remain limited [2]. Recently, the most reliable histopathologic criterion for diagnosing microscopic angioinvasion has been tumor cell infiltration through a vessel wall accompanied by thrombus or a fibrin-tumor complex, or the presence of intravascular tumor cells intermixed with platelet thrombus or fibrin (Fig. 3b) [32, 37]. On the other hand, sinusoidal invasion is variably interpreted either as the presence of tumor cells within thin-walled vascular spaces inside the tumor or, more in line with current guidelines for pathology reporting [2, 32], as the invasion of lymphatic vessels at the periphery of the tumor. The lack of standardized definitional criteria contributes to the equivocal interpretation of sinusoidal invasion and makes it challenging to distinguish true invasion from artifacts caused by surgical specimen handling.
In line with the heterogeneity at macroscopy, ACC displays striking microscopic variability, which is often found in combination within the same tumor, both in terms of tumor architecture and cytological features. The most common pattern is broad trabecular growth, followed by alveolar or large nested, and solid/diffuse arrangements. The presence and extent of a solid or diffuse growth pattern should be noted, as it constitutes one of the criteria in the Weiss scoring system for ACC diagnosis [33]. Less frequently, pseudopapillary and storiform patterns may be observed. Despite this architectural heterogeneity, a unifying feature across all patterns is the loss of the well-organized alveolar architecture characteristic of non-neoplastic adrenal cortex. This architectural disarray serves as a valuable diagnostic clue as it can be demonstrated by the loss of the reticulin framework on silver stain-based histochemistry [5].
Most ACCs are composed of eosinophilic (lipid-poor) tumor cells, which may occasionally exhibit granular cytoplasm. Less frequently, ACC demonstrates clear (lipid-rich) cells, where the lipid content may be diffusely distributed within the cytoplasm or organized in a single vacuole displacing the nucleus, imparting a sort of “signet-ring” appearance. Nuclear atypia, pleomorphism, and hyperchromasia are almost invariably present. Notably, nuclear atypia may also occur in benign adrenal cortical lesions and is therefore a nonspecific feature. In contrast, the presence of one or more centrally located, prominent nucleoli is more characteristic of ACC and constitutes a key diagnostic criterion in the Weiss scoring system. The extent of nuclear pleomorphism can vary significantly within the same lesion and may include bizarre, multinucleated cells or, more rarely, rhabdoid features. In this context, the nuclear features of ACC are generally equivalent to grade 3 (prominent nucleoli visible at 100× magnification) or grade 4 (marked pleomorphism with anaplasia) according to Fuhrman grading criteria for renal cell carcinoma [38].
The presence of coagulative tumor necrosis is another important morphological feature to consider (Fig. 3c). When present, it is typically extensive, broad, and confluent, often exhibiting a comedo-like pattern. However, it may occasionally appear as punctate or focal, which increases the risk of it being overlooked, particularly in cases of suboptimal sampling. In terms of diagnostic and prognostic implications, necrosis is generally assessed as present or absent, whereas no studies have investigated, so far, the possible impact of the evaluation of the extent of necrosis, whenever present, in the characterization of ACC. A main limitation is related to the absence of clear and widely accepted definitions for focal vs. extensive necrosis, at variance with other tumor settings (i.e., sarcomas).
Other two parameters that are strongly associated with malignancy and are integrated in the diagnostic algorithms for ACC are the increased mitotic activity and the presence of atypical mitoses. The cutoff for mitotic index is defined as > 5 mitoses/10 mm^2^ for adults [2] and > 15 mitoses per 4 mm^2^ (20 HPF) for the pediatric patients [39, 40]. However, it is worth noting that in most studies of the available literature on the diagnostic and prognostic impact of mitotic index, mitotic count is expressed in 50 HPF rather than in 10 mm^2^, the latter being a strong recommendation of the last WHO classification scheme only. Therefore, future studies are needed to validate or refine clinically relevant cutoff values of mitotic index expressed in mm^2^. Atypical mitotic figures suggest underlying chromosomal abnormalities such as aneuploidy and are therefore regarded as a hallmark of malignancy, even when only a single, yet unequivocal, atypical mitotic figure is identified (Fig. 3d).
Lastly, tumor stroma may be characterized by intersecting fibrous bands and may display foci of dystrophic calcifications, which can be detected in up to 20% of cases. Lipomatous or myelolipomatous metaplasia can occur, while metaplastic bone formation is rarely seen. It is interesting to note that a lymphocytic inflammatory infiltrate can also be present at tumor periphery or intratumorally. Recently, it has been demonstrated that steroid production in ACC, in particular cortisol secretion as demonstrated by the expression of CYP17A and CYP11B1, significantly interferes with the tumor immune microenvironment, with special reference to the presence of inhibitory Treg lymphocytes [41].
Histological subtypes
In addition to the above-described conventional type of ACC, three histological subtypes are recognized, in the descending order of frequency: oncocytic, myxoid, and sarcomatoid [42] (Fig. 4). It is noteworthy that the WHO classification 5th edition adopts the term “sub-type” instead of the previously used “variant,” aiming to distinguish morphological categories (former) from genetic alterations (latter) [2].Fig. 4. Histological subtypes. The oncocytic subtype is characterized by a predominant diffuse growth pattern (a, 10×) and large eosinophilic cells with abundant granular cytoplasm and nuclear atypia with prominent nucleoli (b, 40×). The myxoid subtype features neoplastic cells arranged in cords, thin trabeculae and microcysts admixed with variable amount of extracellular mucin (c, 10×) and less pronounced cytologic atypia (d, 40×). Lastly, the sarcomatoid subtype is characterized by a storiform architecture (e, 10×) composed by spindle cells featuring sarcomatoid appearance (f, 40×). Conventional ACC component may be present (e, 10×, upper half of the image). In the absence of the conventional component, differential diagnosis with pure sarcomas may be not always possible
These morphological patterns may blend with conventional features to a varying degree, ranging from complete absence to their prevalence, or even complete replacement of the conventional morphology.
The most common is the oncocytic subtype, defined by the presence of oncocytic cells comprising more than 90% of the tumor mass [34, 43]. These cells are large and characterized by their abundant, intensely eosinophilic, granular cytoplasm, which distinguishes them from the eosinophilic cells seen in conventional ACC. Another distinguishing feature of oncocytic ACCs lies in their consistent presentation of prominent nucleoli and a diffuse growth pattern, regardless of their underlying biological behavior.
The myxoid subtype is defined by the presence of a variable amount of extracellular myxoid-like material [44, 45], within which neoplastic cells are arranged in tiny trabeculae, cords, and/or microcysts. Importantly, myxoid changes alone are not diagnostic of malignancy.
Finally, the sarcomatoid subtype represents the least common form of ACC. It is characterized by mesenchymal differentiation in the context of a recognizable cortically derived carcinomatous component [46–48]. In the absence of the latter, these tumors can be indistinguishable from adrenal sarcomas [49, 50] if not for the presence of an even focal adrenal cortical marker expression. Importantly, this subtype has not been reported in the pediatric population.
A critical reappraisal of scoring systems and diagnostic algorithms
The diagnosis of ACC relies on multiparametric scoring systems which variably integrate different histopathological features such as evidence of invasion, architectural and cytological features, mitotic activity, and the presence of necrosis [5, 31, 34, 35, 51, 52] (Table 1). Unfortunately, nearly all of these histopathological parameters are laden with interpretive complexity, posing significant challenges in routine diagnostic practice. Therefore, the integration of multiple algorithms is particularly advisable in cases of adrenocortical lesions where overt clinical or morphological indicators of malignancy are lacking. Table 1. Main scoring/diagnostic algorithms in ACCParameterWeiss scoreModified Weiss (Aubert)Helsinki scoreReticulin algorithmLin-Weiss BiscegliaWieneke/AFIP criteriaApplicabilityAdult ACC (except oncocytic subtype)All adult + pediatric ACCOnly adult oncocytic subtype ACCOnly pediatric ACCCapsular invasion11**--Minor criterion1 (+ 1 extra if extra capsular extension)Angioinvasion1--Additional parameterMajor criterion1 (+ 1 extra if invasion of vena cava)Sinusoidal (lymphatic invasion)1---Minor criterion-Nuclear atypia (grade 3–4^#^)1-----Clear cells < 25%12----Diffuse architecture > 30%1-----Coagulative necrosis115Additional parameterMinor criterion1Mitotic count > 5/10 mm^2^123Additional parameterMajor criterion1 (if > 15/20 HPF)Atypical mitotic figures11--Major criterion1Ki67 index (as %)--Numeric value of Ki67%---Disruption of reticulin framework---Main parameter--Size > 10 cm----Minor criterion1 (if > 10.5 cm)Weight > 200 g----Minor criterion1 (if > 400 g)Cutoff score for Malignancy ≥ 3 ≥ 3 > 8.5Main + 1 of the additional parameters1 major criterionUMP: if 1 or more minor criteria only is present > 3*UMP: 3Benign: 0–2*Main advantagesMostly validated; no special stains requiredNo special stains requiredEasy to assess, good reproducibilityMost validated in oncocytic sub-types; no special stains requiredNo special stains requiredMain disadvantagesPoor reproducibility of some parameters; risk of overestimation for the oncocytic subtype; risk of underestimation for the myxoid subtypeRisk of overestimation for the oncocytic subtype; risk of underestimation for the myxoid subtypeNeeds Ki67 stainRequires reticulin stain; need of larger validation; sites of degeneration may be a pitfallNeeds tumor weight; only applicable to oncocytic adult subtypeNot 100% sensitive nor specific; Needs tumor weightLegend. Numbers refer to points; UMP, uncertain malignant potential; #according to Fuhrman’s grading of renal cell carcinoma
The Weiss system, proposed in 1989, is the most widely adopted and validated algorithm, made up of nine histopathological, purely morphological parameters.
Each parameter accounts for 1 point, for a maximum of 9 points, and malignancy was defined by a score of ≥ 3 points [33]. Given that some of these parameters are highly subjective and poorly reproducible [53] and aiming to increase reliability, a modified Weiss score was proposed by Aubert in 2002. This simplified system eliminated the four least reproducible parameters (angioinvasion, sinusoidal invasion, nuclear atypia, and diffuse architecture) and doubled the weight of other parameters (extent of clear cell and mitotic count), for a maximum of 7 points, with malignancy defined by a score of ≥ 3 points [35]. Both scores are inapplicable in pediatric ACCs, as the specificity of the Weiss score has demonstrated to be low, generally overestimating the malignant potential [40]. Similarly, both scores overestimate malignancy in the adult oncocytic subtype but underestimate malignancy in the myxoid subtype. In fact, in oncocytic neoplasms, 3 Weiss parameters (diffuse growth, eosinophilic cytoplasm, and nucleoli) are invariably present also in oncocytic adenomas, whereas in myxoid cases, the diffuse growth pattern, lympho-vascular invasion, and nuclear atypia may be absent or challenging to evaluate, thereby increasing the risk of underdiagnosing a malignant lesion [45].
To address the diagnostic challenges posed by the oncocytic subtype, a dedicated algorithm, the Lin-Weiss-Bisceglia (LWB) system, was introduced in 2004. This model was specifically designed to overcome the limitations of traditional scoring systems when applied to oncocytic adrenocortical tumors. The LWB algorithm is based on three major and four minor criteria for malignancy, and a diagnosis of malignancy is established when at least one major criterion is present. In contrast, tumors exhibiting at least one minor criterion in the absence of major ones are classified as having uncertain malignant potential (UMP). Key limitations of this system include its applicability exclusively to the oncocytic subtype and its reliance on tumor weight, a parameter not consistently available [34].
More recently, the Helsinki score has emerged as a streamlined and effective diagnostic tool. This algorithm relies on just three parameters: mitotic count, the presence of tumor necrosis, and the Ki-67 proliferative index, specifically measured in the most proliferative area of the tumor, with a final score > 8.5 supporting a diagnosis of ACC [52]. In comparative analyses, the Helsinki score has demonstrated superior predictive accuracy for malignancy over the Weiss system [49], with 100% sensitivity and 99.4% specificity for identifying a metastatic potential. Furthermore, a threshold of 28.5 has been validated as a prognostic indicator of overall survival in a large cohort study [54]. Additionally, the system has been extensively validated across independent cohorts, including both conventional ACC and its histological subtypes [55], as well as in the pediatric population (at the cutoff score of 24) [56].
The reticulin algorithm [31] is predicated on the observation that ACC displays a significant degree of architectural disarray, reflected by qualitative and/or quantitative alterations in the reticulin network (Fig. 5) [37], as demonstrated by silver-based staining methods [31]. For the diagnosis of ACC, the algorithm requires the demonstration of an altered reticulin framework combined with the presence of at least one of three additional parameters, namely necrosis, increased mitotic index, and vascular invasion. The reticulin algorithm has 100% sensitivity and specificity in distinguishing cases coded as benign or malignant by the Weiss system, but it is easier and more reproducible, and its accuracy has been validated in multiple independent cohorts [55, 57], including the pediatric population [56]. Additionally, its potential for objective quantification through computerized morphometric analysis [58] suggests possible future integration into computational pathology–supported diagnostic tools.Fig. 5. Patterns of reticulin stain in ACC. Normal reticulin pattern in normal adrenal cortex features a regular alveolar architecture (a, 20×). In contrast, ACC displays an altered reticulin pattern, manifesting either in form of “qualitative changes” or “quantitative changes”. Qualitative changes (b, 20×) retain the overall reticulin network but show irregular fiber thickness encasing small groups or individual tumor cells. Quantitative changes (c, 20×) are defined by fiber disruption determining a loss of continuity within the reticulin framework
The Wieneke/AFIP Scoring System, introduced in 2003, was specifically designed for the pediatric population [39]. It encompasses nine histological criteria, with a cumulative score exceeding 3 considered indicative of malignancy. Increasing scores correlate with worse overall and disease-free survival outcomes [59]. Despite its utility, this system is limited by its exclusive applicability to pediatric cases and by the incomplete sensitivity and specificity of its criteria.
ACC histopathological tumor grading
According to the WHO classification 5th edition, ACCs should be graded using a two-tiered system based on the mitotic count. ACC is therefore classified as low-grade ACC (mitotic count is ≤ 20 mitoses per 10 mm^2^) or high-grade (mitotic count is > 20 mitoses per 10 mm^2^) [2, 37]. This grading system has originally been proposed by Weiss in 1989 [33] and has since been validated in several adult patients’ cohorts [32, 37, 60] proving to carry a significant prognostic value [33, 37]. Interestingly, recent studies in two independent cohorts have suggested that a cutoff of 10 mitoses per 10 mm^2^ may offer improved prognostic performance in ACC [37, 61], but broader validation is needed.
Importantly, no formal grading system has yet been established for the pediatric population, as an optimal mitotic count threshold for stratification remains to be determined through large-scale clinical studies.
Lastly, although the Ki-67 proliferation index has been shown to have prognostic significance [59], it has not been adopted as a grading tool in the current WHO classification, as such proliferation indices represent continuous variables in tumor biology, rather than fixed/static cutoff points [2].
A practical approach to immunohistochemical markers
Immunohistochemical markers in ACC serve three main purposes: confirming the adrenocortical origin (Table 2), assisting in the distinction between benign and malignant adrenal cortical lesions, and providing prognostic information. Table 2. Immunohistochemical markers of primary adrenocortical originLegend. Markers are color coded: green refers to favorable characteristics, yellow indicates potential issues while red indicated major issues. DDx, Differential diagnosis; PHEO, phaeochromocytoma; NEN, neuroendocrine neoplasms
The former two are discussed below, while the latter is addressed in the dedicated section below.
The most reliable biomarker to confirm the adrenocortical origin is SF1 [62], a nuclear receptor involved in the regulation of steroidogenesis [63]. SF1 exhibits nuclear staining (Fig. 6) and demonstrates excellent diagnostic performance, with up to 100% specificity and 95% sensitivity [64, 65]. However, despite its value, antigenicity may be lost in sub-optimally fixed specimens, and more broadly, the antibodies are not readily available in all centers.Fig. 6. Immunohistochemical markers of adrenocortical origin. SF1 nuclear positivity is the most reliable marker confirming the cortical origin (a, 20×). Other markers of cortical origin, although less specific, include the cytoplasmic expression of Melan A (b, 20×), synaptophysin (c, 20×), α-inhibin (d, 20×), and calretinin (e, 40×). Conversely, cytokeratin (here the AE1/AE3 clone) is usually negative in ACC (f, 40×)
Cytoplasmic markers with lower specificity for adrenocortical origin such as Melan-A, synaptophysin, α-inhibin, calretinin, and D2-40 are more broadly available [62], but their diagnostic performance varies: Melan-A and synaptophysin offer high sensitivity but moderate specificity, α-inhibin shows low sensitivity with moderate specificity, and calretinin and D2-40 are both the least sensitive and specific among them. It should be emphasized that some of the abovementioned markers may also be expressed by neoplasms that closely mimic ACC: Melan-A can be expressed by melanoma [66], PEComa [67], and renal cell carcinoma (RCC) [68]; synaptophysin is positive in paragangliomas [62] and neuroendocrine neoplasms; α-inhibin is also positive in a subset of paragangliomas and various non-adrenal carcinomas [69]. In such contexts, employing a panel of adrenocortical markers alongside lineage-specific immunohistochemical stains tailored to the differential diagnosis can enhance diagnostic accuracy and reduce the risk of misdiagnosis. A case of primary adrenal malignant PEComa is illustrated in Fig. 7, as an example of an ACC mimicker.Fig. 7. Intra-adrenal PEComa mimicking ACC. The intra-adrenal lesion (a, 5×; bottom left, peritumoral adrenal cortex) is composed of epithelioid eosinophilic cells arranged in a vaguely trabecular architecture (b, 40×). Metastasis to a periadrenal lymph node is shown in (c, 10×). In contrast to the adjacent normal adrenal cortex, the tumor shows the loss of SF1 expression (d, 25×). The present case was also negative for Melan A (e, 25×). Further immunohistochemical analyses revealed diffuse cathepsin K positivity (f, 25×) and a moderately low proliferative index, with Ki-67 staining around 10% (g, 25×)
If cortical origin is immunohistochemically confirmed, the presence of invasive growth or high-grade features warrants the diagnosis of ACC. Conversely, in front of low-grade features, the diagnosis of ACC should be challenged with that of adrenocortical adenoma and may require ancillary biomarkers [37, 62]. To this end, in addition to the histopathological features composing multiparametric scoring systems and the previously discussed reticulin silver stain, some immunohistochemical markers of malignancy have been proposed in adrenal cortical tumors. Among them, insulin-like growth factor 2 (IGF2) immunostaining can be utilized. With a juxtanuclear granular staining pattern, IGF2 has proven to be a specific marker for ACC ranking as the most reliable ancillary tool for distinguishing ACC from adenoma [2]. Additionally, p53 could also be employed, as the altered expression of this marker supports the diagnosis of ACC, as also discussed below. However, as this alteration is more typical of high-grade ACC, its diagnostic accuracy in the context of low-grade features is notably low [62].
Pathological, immunohistochemical, and molecular prognostic markers
From a pathological standpoint, numerous histopathological features have been recognized as prognostically relevant. Capsular invasion has been recognized as an independent risk factor for mortality, and angioinvasion is emerging as one of the most powerful prognostic indicators in ACC [37], highlighting the critical importance of accurate identification of these features. More broadly, positive surgical margins were found to be an independent predictor of both shorter overall survival (p = 0.04) and recurrence-free survival (p = 0.03)[70], reinforcing earlier evidence demonstrating a significantly increased risk of mortality associated with margin involvement in multivariable analysis (p < 0.0001) [71]. Lastly, necrosis has generated particular interest as its presence has been shown to adversely affect both overall survival (p = 0.05) and disease-free survival (p < 0.001), emerging as the strongest adverse prognostic factor within the Weiss scoring system [72].
Concerning immunohistochemical biomarkers, the most relevant is the Ki-67 proliferation index [59, 73] (Fig. 8), although a consensus on prognostic cutoff values has yet to be established. In this context, the development of diagnostic algorithms, whether based on manual counting or automated image analysis, may improve the standardization of its assessment [74], potentially helping to bridge this gap in the future. This would be particularly valuable given the index’s utility in guiding decisions regarding adjuvant mitotane therapy [22]. However, a main limitation in the application of Ki-67 in clinical practice is represented by low reproducibility, with special reference to the interpretation of the results that leads to poor inter-observer agreement [75]. The use of digital image tools has been claimed to represent a possible solution to implement reproducibility, but it needs appropriate settings, in particular to reduce the risk of overestimating the proliferation index [74].Fig. 8. Heterogeneity of Ki-67 index in ACC. Ki-67 proliferation index may be lower than 10% in some cases (a, 10×) but in most cases is elevated (b, 10×)
Another proliferative marker, phosphohistone H3 (PHH3), has been implied to improve the accuracy of mitosis identification [61], whose important prognostic role has been discussed previously. However, its utility has not yet been fully validated.
P53 and β-catenin are recognized as key translational prognostic biomarkers, as altered expression of these proteins is frequently observed in carcinomas associated with high-risk molecular profiles. Aberrant p53 staining presents as either diffuse nuclear expression or complete absence, while altered β-catenin expression is marked by diffuse nuclear expression [62] (Fig. 9). Additional markers linked to poorer clinical outcomes include high expression of SF1 [62, 64] and loss of ATRX and ZNRF3 expression [76].Fig. 9. Immunohistochemical stains as surrogate molecular markers in ACC. Altered p53 expression presented as overexpression (a, 20×); altered β-catenin expression, as evidenced by aberrant nuclear positivity (b, 20×); mismatch repair deficiency (MMRd) evidenced by the loss of MSH6 protein expression, with intact internal control (c, 20×) and preserved MSH2 (d), MLH1 (e, 20×), and PMS2 (f, 20×) expression
Moreover, immunohistochemical evaluation of mismatch repair (MMR) proteins (Fig. 9) and SDHB can aid in identifying underlying germline alterations associated with Lynch syndrome and SDH-related familial paraganglioma syndrome, respectively. Consequently, the application of these markers is recommended in all apparently sporadic cases of adrenocortical carcinoma [62]. Additionally, MMR proteins, together with PD-L1 immunohistochemistry, could help identify patients with susceptibility to immune-enhancing therapies [77].
Finally, the prognostic role of CYP11B1 and CYP11B2, known as immunohistochemical markers of steroid-secreting adrenocortical neoplasms [78] remains controversial in ACC [79].
Recent transcriptomic [80] and pan-genomic [27] studies have increasingly underscored the prognostic potential of molecular characterization in adrenocortical carcinoma (ACC). For instance, alterations in the TERT gene have been linked to unfavorable clinical outcomes, including metastatic progression and disease-specific mortality [81]. Similarly, methylation profiling has emerged as a valuable tool for predicting prognosis [26]. In this context, dysregulation of microRNAs, such as downregulation of miR-195 and overexpression of miR-483-5p, as well as hypermethylation of the GO/G1 Switch 2 (G0S2) gene, has all been associated with poorer outcomes and increased mortality risk [82, 83]. Furthermore, RRM1 gene expression has gained attention as a predictive biomarker for response to adjuvant mitotane therapy in ACC while elevated CYP2W1 mRNA levels have been correlated with improved survival in patients receiving mitotane treatment [84].
A schematic overview of the key characteristics of the principal immunohistochemical and molecular biomarkers is shown in Table 3. Table 3. Prognostic immunohistochemical and molecular biomarkers in ACCLegend. Light gray boxes indicate immunohistochemical markers with stronger validation and are thus the most recommended; MMRd, deficient mismatch repair protein function; *, no specific PD-L1 scoring criteria or cut-offs have been officially established to date
Synoptic standardized pathology reporting
The pathological evaluation of ACC remains challenging, complex, and potentially ambiguous, and a standardized approach to the pathological evaluation of ACC would significantly enhance risk stratification for individual patients and would enable robust multinational translational research [32]. To this end, in 2021, the International Collaboration on Cancer Reporting (ICCR) convened an expert panel to review the pathological reporting of ACC and subsequently established a standardized dataset for ACC reporting, now available on the ICCR website (https://www.iccr-cancer.org/datasets/published-datasets/endocrine/adrenal-cortex/).
The dataset subdivides elements into core and non-core. Briefly, the core elements refer to data points deemed essential for clinical management, staging, or prognostication, and for which there is unanimous consensus among the expert committee. In contrast, non-core elements may hold clinical relevance but lack consistent validation or widespread implementation in routine patient management. The application of this scheme is strongly encouraged to implement standardization of pathological reporting and increased diagnostic reproducibility.
All these elements are summarized in Table 4. Table 4. International Collaboration on Cancer Reporting (ICCR) dataset for pathology reporting of ACC. Data adapted from (https://www.iccr-cancer.org/datasets/published-datasets/endocrine/adrenal-cortex/)Core elementsNon-core elementsClinicalClinical information (e.g., symptoms, functionality, syndromes, prior therapy)Operative procedure (open or laparoscopy)Type of specimen submitted (also specimen other than adrenal gland should be identified)Tumor siteMacroscopicalSpecimen integrity (intact or fragmented)Tumor dimensions (largest single dimension)Additional two dimensionsTumor weight (after adipose tissue and other organs are removed)MicroscopicalHistological tumor type (according to the WHO)Extent of invasion (invasion of extra-adrenal adipose tissue or nearby organs)Tumor architecture (trabecular, alveolar, nested or diffuse)Clear (lipid-rich) cellsCapsular invasionLymphatic (sinusoidal) invasionVascular invasionAtypical Mitotic FiguresCoagulative tumor necrosisExtent of necrosisNuclear gradeMitotic count and histological tumor gradeKi67 proliferation index (measured on the area with the highest mitotic count)Margin statusDistance of the tumor to the closest marginLymph node statusExtra-nodal extensionHistologically confirmed distant metastasisPathologic staging (UICC/AJCC)Multifactorial scoring systemsAncillary studies (reticulin, SF1, NGS)Coexisting adrenal pathology (e.g., adenoma)Legend. UICC, Union for International Cancer Control; AJCC, American Joint Committee on Cancer; NGS, next-generation sequencing
Potential utility and limitations of artificial intelligence application in the field of ACC
In the field of ACC, as in many other medical fields, the role of artificial intelligence (AI) is rapidly expanding.
Considering that the CT scan represents a mandatory diagnostic tool used in patients with a clinical suspicion of adrenal mass, it is unsurprising that it has received considerable interest. In 2022, a Japanese retrospective single-center study used two methods (U-Net architecture and region-based convolutional neural network) to develop AI models to detect and classify adrenal masses. Although AI assistance was associated with improved sensitivity for less experienced radiologists, for an experienced physician, AI suggestion seemed to hamper performance [85]. Two preliminary works, both of which incorporated radiomics features, were presented. A retrospective Chinese multi-institutional study extracted radiomics features from different phases of contrast-enhanced CT images from 158 patients and developed an interpretable radiomics model which had superior diagnostic performance compared to two experienced radiologists (AUC model 0.92 vs. AUC radiologist 1 0.79, AUC radiologist 2 0.63) [86]. A larger retrospective European study analyzed 794 adrenal masses using texture analysis on unenhanced CT scans and showed that a radiomic-based DL algorithm was highly accurate in predicting the presence of malignant adrenal masses and specifically performed well in predicting ACC (AUC = 0.933, F1-score = 0.318, sensitivity = 96.4%, specificity = 83.9%) [87].
To the best of our knowledge, very few AI tools specifically developed for the diagnosis of ACC have been published. We found a single very recent study in the literature that applied pathomics analysis to ACC. In this study, a specific signature based on 5 features (related to cell density, chromatin characteristics, and staining intensity) was developed and integrated with clinical characteristics into a nomogram that proved to have prognostic impact in ACC [88].
The application of deep learning techniques to whole slide images in the context of ACC has likely been hindered by the rarity of the disease and consequent limited availability of digitized ACC histology slides, significant histological heterogeneity, which complicates model development, and intrinsic diagnostic difficulties with varying diagnostic interpretation even among experts which translate into an inability to obtain enough reliable ground truth annotations.
More broadly, ethical challenges persist in deploying these systems, as they have been developed in specific populations and this potentially may compromise model accuracy in other populations. Lastly, the limited explainability of current AI systems remains a key barrier to their clinical adoption. Addressing the “black box” nature of deep learning algorithms will require not only technical advances in interpretable modeling, but also sustained interdisciplinary collaboration between pathologists, computer scientists, and regulators, to ensure that future AI tools will be scientifically robust and clinically trustworthy.
The reference list from the paper itself. Each links out to its DOI / PubMed record.