Genomic characterization of Listeria monocytogenes isolated from small ruminants in integrated crop-livestock systems
Sejin Cheong, Joanna G. Rothwell, Cory Schlesener, Bart C. Weimer, Craig Miramontes, Richard V. Pereira, Paulo Pagliari, Alda F. A. Pires

TL;DR
This study examines Listeria monocytogenes strains from sheep and goats in crop-livestock systems, finding potentially dangerous strains in the environment.
Contribution
The study provides genomic insights into L. monocytogenes from small ruminants in integrated systems, linking them to human disease outbreaks.
Findings
Most isolates belonged to lineage I and clonal complexes CC1 or CC554, associated with human listeriosis outbreaks.
Isolates carried virulence genes like LIPI-1 and agrA, linked to biofilm formation and environmental survival.
All isolates were susceptible to antibiotics used for treating listeriosis in humans and ruminants.
Abstract
Listeria monocytogenes (L. monocytogenes), a foodborne pathogen shed by asymptomatic ruminants, poses a contamination risks in integrated crop-livestock systems (ICLS), where ruminants are introduced to graze cover crops prior to replanting fresh produce in a field. As a follow-up study, we conducted whole genome sequencing of 30 L. monocytogenes isolates obtained from sheep and goat fecal and soil samples collected during our previously published ICLS field trial studies (2019—2022) at organic farms in California and Minnesota. One goat fecal isolate was genetically identical to one soil isolate collected at seven days post-grazing. Most isolates (28/30, 93.3%) belonged to lineage I, specifically to serogroup IVb or IVb-v1, and were classified as CC1 or CC554, both clonal complexes previously associated with human listeriosis outbreaks. The majority of isolates harbored virulence…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
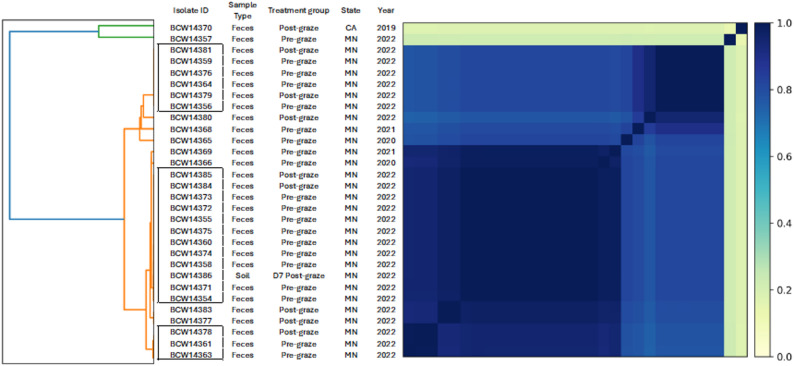 Figure 1
Figure 1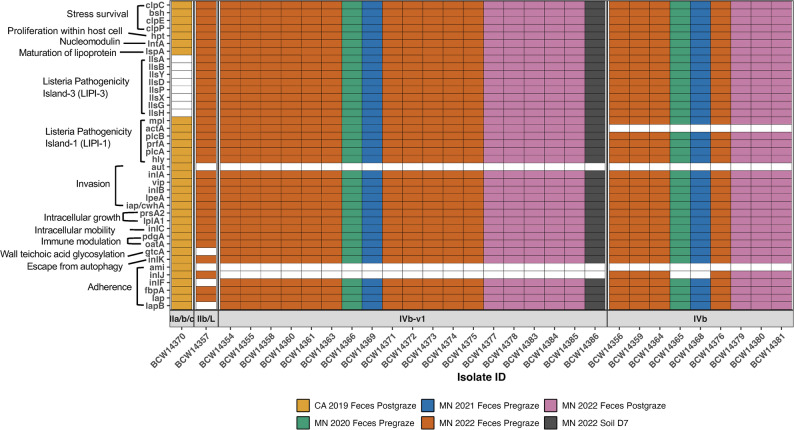 Figure 2
Figure 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsListeria monocytogenes in Food Safety · Salmonella and Campylobacter epidemiology · Bacillus and Francisella bacterial research
Introduction
Listeria monocytogenes (L. monocytogenes) is a foodborne pathogenic bacterium that can survive and persist in diverse environments, including feces of animals, soil, and vegetation [1–3]. Listeriosis in humans remains a significant public health and clinical concern due to its fatal outcomes such as septicemia, central nervous system infections, and abortion [4]. Most reported human cases have been linked to the consumption of contaminated dairy or meat products [4–6]. However, asymptomatic shedding of L. monocytogenes by healthy animals, including cattle, sheep, and goats, has been documented, contributing to environmental contamination on farms [7, 8]. Although infections linked to fresh produce contaminated via animal feces and soil in vegetable farms are relatively rare, L. monocytogenes presence in growing environments represents a significant food safety concern [6, 9]. Indirect contamination of L. monocytogenes from livestock to fresh produce has been associated with human outbreaks; for instance, sheep manure was identified as the source of L. monocytogenes outbreak in cabbage used for coleslaw [10].
With the increasing application of whole genome sequencing (WGS) to trace transmission routes of L. monocytogenes for surveillance and outbreak investigation, understanding potential sources of contamination in complex farm environments has become more feasible [5, 11–13]. Previously, serotyping (e.g., 1/2a, 1/2b, 4b) or serogrouping (e.g., IVb, IVb-v1, IIa, IIb) based on specific antisera has been performed over decades despite its relatively low discriminatory power [14]. Recently, however, phylogenetic analyses including multilocus sequence typing (MLST) using seven housekeeping gene fragments and WGS have enabled identification of four distinct lineages (I–IV), and further subdivided into clonal complexes (CCs) and sequence types (STs) [15]. Each ST represents a unique allelic profile of the seven housekeeping genes, and CCs group STs that differ by a single locus [16], thereby enabling more detailed strain comparisons. For example, Dreyer et al. (2015) investigated the transmission of L. monocytogenes between sheep and the farm environment by performing three genetic subtyping methods and confirmed that identical strains were isolated from sheep, soil, and water tanks in a farm [17]. Furthermore, genomic analyses including virulence genes, antimicrobial resistance genes, and mobile genetic elements enable the identification of the genetic diversity and evolution of L. monocytogenes, which may explain its wide distribution in the environment and among different hosts [13, 18]. However, the extent of fecal shedding of L. monocytogenes from small ruminants and its persistence in agricultural farm environments has not been extensively investigated [9, 15, 19]. In particular, limited studies have focused on asymptomatic small ruminants [7, 8, 20], although some studies have examined small ruminant cases with clinical symptoms, such as neurological disorders [21–23].
As one of the regenerative farming practices, integrated crop-livestock systems (ICLS) involve grazing small ruminants on cover crops grown between fresh produce planting cycles [24, 25]. While raw manure can serve as an organic fertilizer, it poses a potential risk of transferring L. monocytogenes from asymptomatic grazing animals to fresh vegetable growing environments [26, 27]. In our previous four-year ICLS field trials (2019—2022), sheep and goats were directly introduced into fields to graze cover crops prior to planting tomatoes, spinach, and cucumbers [28, 29]. L. monocytogenes was detected in 0.5% (1/220) of sheep fecal samples from California (CA), and in 21.9% (35/160) of goat fecal samples and 0.2% (1/540) of soil samples collected in Minnesota (MN). L. monocytogenes was rarely found in soil and was not detected in any vegetable samples in our previous studies. The single positive soil sample detected in MN may have originated from the small ruminants. However, no high-resolution methods were applied at that time to confirm this.
To build upon the findings of our previously published studies [28, 29], the present study conducted a follow-up investigation to further characterize 30 L. monocytogenes isolates obtained from the previous ICLS field trials. Specifically, this study aimed to 1) assess the genetic similarity of L. monocytogenes isolated from fecal and soil samples detected, and 2) characterize L. monocytogenes isolates from the feces of asymptomatic sheep and goats used for grazing, using WGS data. Given the limited research on asymptomatic small ruminants, this study contributes to filling a knowledge gap regarding L. monocytogenes isolates from healthy small ruminants in agricultural settings.
Materials and methods
Sample collection and L. monocytogenes isolation methods
A total of 30 L. monocytogenes isolates including 28 from goat feces, one from sheep feces, and one from soil, were obtained during crop-livestock integration field trials (2019—2022) in CA and MN, as described in our two previous studies [28, 29]. Of the 28 isolates from goat feces, 24 isolates were obtained in 2022, 16 from pre-graze and eight from post-graze treatment groups (Table 1). The two previous studies were field trials with a randomized complete block design with four replicates. Three treatments (fallow as a control, cover crop tilled without grazing, cover crop grazed by sheep or goats) were randomly assigned to each block. Following the cropping sequence generally adopted by each region, cover crop mix was seeded (November in CA, August/September in MN), and grazing was started when the cover crop height was approximately 25 cm in the grazed treatment plots. Depending on the size of plots (4.5 × 121.9—236.2 m in [28] and 4.5 × 22.9 m in [29] per one paddock with four replicates) and amount of biomass in each year, 25—120 sheep in CA and 115—170 goats in MN grazed the cover crops. Fecal samples were collected both before grazing from the barn or trailer floors and after grazing from the plots immediately after sheep or goat were removed from the grazed plots. Briefly, to isolate L. monocytogenes, fecal (10 g) and soil (30 g) samples were enriched in Listeria Enrichment Broth (90mL and 270mL, respectively) (Neogen Culture Media, Lansing, MI), followed by Immunomagnetic separation (IMS) [30]. A 30 μL aliquot of the IMS washed bead product was plated on Brillance Listeria Agar (BLA) with selective supplements (Oxoid, Hants, United Kingdom), while 100 μL of the IMS product was also added into Fraser broth (BD, Sparks, MD). Both media were incubated for 48 h at 37 °C. After the sequential isolation process, presumptive positive colonies (blue colonies with a halo) were streaked onto Tryptic Soy Agar (TSA). L. monocytogenes were confirmed via conventional PCR targeting the hylA gene [31]. Isolates from each positive sample were preserved in 15% glycerol with Tryptic Soy Broth at −80 °C. For further regrowth for DNA extraction and antimicrobial susceptibility testing, one isolate was selected from each sample to ensure that each isolate corresponded to an individual sample.Table 1. Isolates of L. monocytogenes (n = 30) from crop-livestock integration field trials (2019—2022). Sheep were used as grazing animals in California (CA), while goats were used in Minnesota (MN)Isolate IDSample TypeTreatment groupStateYearSerogroup (SG)SequenceType (ST)ClonalComplex (CC)LineageBCW14370FecesPost-grazeCA2019IIa,IIb, or IIc918CC918IIBCW14357FecesPre-grazeMN2022IIb or L1965CC1283IIIBCW14381FecesPost-grazeMN2022IVb4CC4IBCW14359FecesPre-grazeMN2022IVb4CC4IBCW14376FecesPre-grazeMN2022IVb4CC4IBCW14364FecesPre-grazeMN2022IVb4CC4IBCW14379FecesPost-grazeMN2022IVb4CC4IBCW14356FecesPre-grazeMN2022IVb4CC4IBCW14380FecesPost-grazeMN2022IVb4CC4IBCW14368FecesPre-grazeMN2021IVb219CC4IBCW14365FecesPre-grazeMN2020IVb1CC1IBCW14369FecesPre-grazeMN2021IVb-v1554CC554IBCW14366FecesPre-grazeMN2020IVb-v1554CC554IBCW14385FecesPost-grazeMN2022IVb-v1554CC554IBCW14384FecesPost-grazeMN2022IVb-v1554CC554IBCW14373FecesPre-grazeMN2022IVb-v1554CC554IBCW14372FecesPre-grazeMN2022IVb-v1554CC554IBCW14355FecesPre-grazeMN2022IVb-v1554CC554IBCW14375FecesPre-grazeMN2022IVb-v1554CC554IBCW14360FecesPre-grazeMN2022IVb-v1554CC554IBCW14374FecesPre-grazeMN2022IVb-v1554CC554IBCW14358FecesPre-grazeMN2022IVb-v1554CC554IBCW14386SoilD7 Post-grazeMN2022IVb-v1554CC554IBCW14371FecesPre-grazeMN2022IVb-v1554CC554IBCW14354FecesPre-grazeMN2022IVb-v1554CC554IBCW14383FecesPost-grazeMN2022IVb-v1554CC554IBCW14377FecesPost-grazeMN2022IVb-v1554CC554IBCW14378FecesPost-grazeMN2022IVb-v1554CC554IBCW14361FecesPre-grazeMN2022IVb-v1554CC554IBCW14363FecesPre-grazeMN2022IVb-v1554CC554I
Whole-genome sequencing
Isolates stored in 15% glycerol cryotubes were plated on TSA for regrowth and incubated overnight at 37 °C. A 10 μL loop of colonies was inoculated into Mueller–Hinton Broth (MHB) (MilliporeSigma, Carlsbad, CA) and incubated overnight. A total of 1.5 mL of MHB culture was transferred into two microcentrifuge tubes and centrifuged at 13,000—16,000 × g for 5 min. Genomic DNA was extracted from the resulting pellets using the Wizard genomic DNA purification kit (Promega Corporation, Madison, WI, USA), following the manufacturer’s instructions for gram positive bacteria, with slight modifications. Briefly, 20 μL of lysozyme and mutanolysin were substituted at the lytic enzyme addition step and following the first incubation step 15 μL of proteinase K was added and incubated at 65 °C for 10 min. The resulting DNA pellet at the end of protocol was rehydrated with Tris HCl solution overnight at 4 °C. DNA concentration (ng/μL) and purity (A260/280 ≥ 1.8 and A260/A230 ≥ 2.0 used as thresholds for the next step) were quantified using a Nanodrop One UV–Vis Spectrophotometer (ThermoScientific, Waltham, MA, USA). Genomic DNA integrity was assessed using TapeStation (Agilent 4200, Santa Clara, CA, USA) before storage at −20 °C until WGS [32, 33].
WGS was conducted following the established 100 K Pathogen Genome Project protocol [34–36]. Sequencing libraries were prepared from high-quality genome DNA through enzymatic fragmentation and size selection (average 450 bp inserts), then sequenced using the Illumina HiSeq X platform (San Diego, CA, USA). Raw sequence data with all accession numbers on the NCBI database are available in the Supplementary Material.
Genome assembly and analyses
Whole genome sequence data were processed with Trimmomatic (version 0.39) to remove low-quality sequence and sequencing adapters [37], using settings “PE ILLUMINACLIP: < adapters > :2:40:15 LEADING:2 TRAILING:2 SLIDINGWINDOW:4:15 MINLEN:50” with program provided Illumina adaptor sequences (“https://github.com/timflutre/trimmomatic/tree/master/adapters”, accessed August 1 st, 2022). Quality of sequence data was assessed by FastQC (version 0.12.1) [38], and samples were removed from analysis if they failed any of the following modules 'Adapter Content', 'Per base sequence quality', 'Per sequence quality scores', and 'Sequence Duplication Levels'.
Contamination by PhiX internal sequencing standard was searched for by Kraken2 (version 2.1.3) [39] and if found, removed by read alignment with Bowtie2 (version 2.5.3) [40], both using the Illumina PhiX reference sequence (https://webdata.illumina.com/downloads/productfiles/igenomes/phix/PhiX_Illumina_RTA.tar.gz, accessed August18, 2020). Microbial taxonomy and contamination were assessed with Kraken2 and Bracken (version 2.9) [41]. Kraken2 microbial database was constructed using the microbial genomes from NCBI RefSeq database (Kraken2 program preset reference libraries archaea, bacteria, viral, fungi, protozoa, and UniVec_Core, downloaded/constructed August 2nd, 2023); Bracken database was built off Kraken2 microbial database by standard protocol, using the parameters of k-mer size = 35 and read size = 150. Contamination seen by Kraken2/Bracken was defined as > 5% of reads matching another species; samples identified as Listeria monocytogenes were used for analysis.
Genome assemblies were constructed with Shovill (version 1.1.0) [42] using the default options with the SPAdes assembler [43]. Genome assembly quality was reviewed with CheckM (version 1.2.2), using the “lineage_wf” workflow [44]. Each assembly’s depth of coverage was measured with Mosdepth (version 0.3.6) [45], using the “fast-mode” setting, and Bowtie2 and Samtools (version 1.19.2) for trimmed reads alignment to assembly preparation [46]. Quality control cutoffs for inclusion in the analyses were CheckM: > 90% estimated completeness, < 6% estimated contamination, within range 2.5–3.5 Mbases for assembly size, < 300 contigs; Mosdepth: > 30 × mean depth of coverage.
Whole genome similarity comparisons were performed using Sourmash (version 4.8.6) with a K-mer size of 31 [47], and a Sourmash heatmap plot was created with a scale of 100,000 k-mers per megabase (scaled sketch size of 10) to assess the genomic relatedness of each L. monocytogenes isolates.
In silico genomic characterization of L. monocytogenes isolates - multilocus sequence typing, virulence associated genes, stress survival islets, biofilm formation related genes, antimicrobial resistance (AMR) genes, and plasmid profiles
Serogroups (SGs) were manually assigned according to the Doumith’s scheme based on the presence/absence pattern of specific marker genes—lmo0737, lmo1118, ORF2110, ORF2819, and prs identified in each assembled genome [48]. Multilocus sequence typing (MLST) was performed using seven housekeeping genes (abcZ, bglA, cat, dapE, dat, ldh, and lhkA) to assign sequence types (STs) with the “mlst” program (version 2.23.0) [49] using “listeria_2” schema of the PubMLST database [50] (database packaged with program, updated 2022 Oct 28). Clonal complexes (CCs) and lineages were inferred from MLST profiles using the L. monocytogenes database hosted by the Institut Pasteur. Genes of interest further listed were searched for in genome assemblies by ABRicate (version 1.0.1) [51], with a standard set of databases (downloaded 2023 Nov 4). Virulence-associated genes were identified by screening against the Virulence Factor Database (VFDB) [52]. The presence of stress survival islets and the sigB (lmo0895) gene was assessed to examine stress tolerance potential, while biofilm formation genes (agrA, agrC, and luxS), potentially associated with soil attachment and adaptation, were identified using the L. monocytogenes database from the Institut Pasteur. AMR genes were detected using multiple databases: ARG-ANNOT [53], the Comprehensive Antibiotic Resistance Database (CARD) [54], MEGARes [55], the NCBI AMR database [56], and ResFinder [57]. The presence of plasmids was assessed using PlasmidFinder [58].
Phenotypic antimicrobial susceptibility testing
Isolates preserved in 15% glycerol cryotubes were plated onto the TSA for resuscitation and BLA to assess potential contamination, followed by overnight incubation at 37°C. L. monocytogenes colonies from TSA plates were submitted to the California Animal Health and Food Safety (CAHFS) Laboratory for antimicrobial susceptibility testing. Testing was conducted using the broth microdilution method with the Sensititre Vet Bovine/porcine plate (BOPO7F) (Thermo Fisher Scientific, Wilmington, DE). As the official breakpoints used to define the susceptibility and resistance profiles of L. monocytogenes are limited to penicillin, ampicillin and trimethoprim–sulfamethoxazole in the Clinical and Laboratory Standards Institute (ICLS M100 ED33) and the European Committee on Antimicrobial Susceptibility Testing (EUCAST), interpretations for clindamycin and gentamicin were made using breakpoints established for Staphylococcus spp., recommended by previous studies [59, 60]. For the other antimicrobials without any breakpoint established, minimum inhibitory concentrations (MIC) were reported.
Statistical analysis
Descriptive statistics were used to summarize the distribution of MLST types, virulence factors, stress survival islets, biofilm formation, AMR genes, and plasmid presence across all isolates. Since pre- and post-graze fecal samples were not collected from the same individual animals, Fisher’s exact test was performed to evaluate the differences in the presence of virulence-associated genes between the treatment groups (pre- and post-graze), with significance set at p < 0.05. A heatmap was generated in R (v.4.3.0) with the ggplot2 package to visualize the distribution of virulence-associated genes.
Results
MLST typing and whole genome comparison of isolates
The draft genome consists of 22—60 contigs with a coverage depth of 95 ×—845 × and is estimated > 99% complete based on the quality control. WGS of L. monocytogenes (30 isolates) identified six STs and CCs by MLST (Table 1). Most isolates (28/30, 93.3%) belonged to lineage I, specifically to serogroup IVb or IVb-v1, and were assigned to CC1 (ST1), CC4 (ST4 or ST219), and CC554 (ST554). The hypervirulent CC1 and CC4 were detected in goat fecal samples collected both pre- (6 isolates) and post-graze (3 isolates) treatments across 2020—2022. In contrast, one isolate identified from a sheep fecal sample belonged to lineage II and was assigned to CC918 (ST918), showing a distinct genetic profile compared to goat fecal isolates.
WGS comparisons identified three clusters of isolates, each displaying complete pairwise genomic similarity (Jaccard index = 1) (Fig. 1). Two of these clusters were identified as CC554 (ST554), the most prevalent CCs/STs. One soil-derived L. monocytogenes isolate (isolate ID: BCW14386), collected at 7 days after goat grazing of the field, showed identical genomic similarity to pre- and post-graze goat fecal isolates collected in the same year. Although this soil isolate was obtained from a non-grazed treatment plot, adjacent to the grazed plot in our previous field trial, its genetic similarity suggests potential transfer from the grazed plot. Interestingly, two pre-graze fecal isolates collected in 2020 (Isolate ID: BCW14366) and 2021 (Isolate ID: BCW14369) showed high genomic similarity to the isolates identified in 2022, with all classified as CC554 (ST554).Fig. 1. Whole-genome similarity heatmap of L. monocytogenes isolates (n = 30) collected from crop-livestock integration field trials (2019—2022). The heatmap visualizes pairwise genomic similarities based on the Jaccard similarity index with the color gradient on the right indicating the relative degree of similarity (0–1)
Profiles of virulence-associated, stress tolerance, and biofilm-related genes in L. monocytogenes isolates
The profiles of virulence-associated genes in L. monocytogenes isolates were generally consistent within each serogroup (Fig. 2). Most isolates, except for the nine isolates classified as serogroup IVb, contained the entire Listeria Pathogenicity Island-1 (LIPI-1), which includes the prfA, plcA, plcB, actA, mpl, and hly genes. A key difference between isolates belonging to serogroups IVb and IVb-v1 was the presence or absence of the inlJ or actA genes. The actA gene, a component of LIPI-1, encodes the actin assembly-inducing protein, which facilitates intercellular mobility and cell-to-cell spread during infection [61]. The InlJ gene encodes a fibronectin-binding protein associated with bacterial adherence [62]. Additionally, all 29 isolates identified from goats and soil contained Listeria Pathogenicity Island-3 (LIPI-3), which encodes listeriolysin S and comprises eight genes: llsA, llsG, llsH, llsX, llsB, llsY, llsD, llsP. However, one isolate from sheep did not have all LIPI-3 genes.Fig. 2. Distribution of virulence-associated genes in L. monocytogenes isolates categorized by state, year, and treatment (pre-/post- graze) group (indicated by color) and serogroup (grouped in box), based on Virulence Factor Database analysis. Absence of color indicates that the gene was not detected
Analysis of isolates by pre- and post-graze treatment groups revealed that most isolates in each group belonged to serogroup IVb-v1 and carried all the LIPI-1 and LIPI-3 genes. Specifically, 65% (13/20) in the pre-graze and 55.6% (5/9) in the post-graze treatment groups contained these virulence factors. No statistical difference was observed between pre- and post-graze fecal samples regarding the presence of the inlJ or actA genes.
All isolates contained stress survival genes listed in the VFDB, including clpC, clpE, and clpP (encoding stress proteins), and bsh (related to bile salt hydrolase production) (Fig. 2). Interestingly, one isolate from sheep feces carried stress survival islet 1 (SSI-1), comprising five genes – lmo0444, lmo0445, lmo0446 (pva), lmo0447 (gadD1), and lmo0448 (gadT1). In contrast, none of the isolates from goats or soils contained these SSI-1 genes, which are known to confer tolerance to environmental stress such as acid, salt, and high osmolality. Additionally, the master regulator sigB gene, involved in stress survival gene regulations, was present in all isolates, except one (BCW14357). Regarding biofilm-associated genes, agrA, agrC, and luxS were detected in all the isolates except BCW14357, which lacked agrC but carried agrA and luxS.
AMR genes and AMR phenotypes
All 30 L. monocytogenes isolates shared identical antibiotic resistance gene profiles, including fosX (fosfomycin), mprF (defensin), Lin (lincomycin), lmo0919_fam (lincomycin), norB (fluoroquinolone), and MDrL. The MDrL is associated with multidrug efflux mechanisms in L. monocytogenes, facilitating the export of macrolide-based antibiotics and cefotaxime [63]. The phenotypic antimicrobial susceptibility testing results are summarized in Table 2. According to CLSI (≤ 2µg/mL; susceptible) or EUCAST (≤ 1µg/mL; susceptible) breakpoints, all isolates were susceptible to ampicillin or penicillin. Due to the presence of a single MIC range on the testing plate, the susceptibility of TMS (Trimethoprim/Sulfamethoxazole) could not be determined. When assessed the other antibiotics using CLSI breakpoints for Staphylococcus spp., all isolates demonstrated intermediate susceptibility (by CLSI; ≥ 4µg/mL; resistant) or resistant (by EUCAST; ≥ 0.25µg/mL; resistant) to clindamycin and were susceptible to gentamycin (by CLSI; ≤ 4µg/mL; susceptible, by EUCAST; ≤ 2µg/mL; susceptible).Table 2. Phenotypic antimicrobial susceptibility testing results with minimum inhibitory concentrations (MIC) of 30 L. monocytogenes isolates. Antibiotics in blue represent those with established breakpoints by CLSI or EUCAST, but the other remaining antibiotics are either mostly used in veterinary medicine or lack defined breakpointsNumber of isolates with MIC (µg/mL) (out of 30 isolates in total)Antibiotics ≤ 0.1250.250.51248163264128 ≥ 256MIC rangeAmpicillin91830.25–16Ceftiofur^^4260.25–8Chlortetracycline^^12630.5–8Clindamycin^+^1290.25–16Danofloxacin^^300.12–1Enrofloxacin^^3270.12–2Florfenicol^^2280.25–8Gentamicin + 301–16Neomycin304–32Oxytetracycline^^6240.5–8Penicillin641820.12–8Sulphadimethoxine^^33256Spectinomycin12818–64Tiamulin^^1291–32Tilmicosin^^10204–64TMS (Trimethoprim/Sulfamehtoxazole)302/38Tulathromycin^^308–64Tylosin^^2910.5–32^^Antibiotics are used in veterinary medicine for livestock in the US^+^Breakpoints established for Staphylococcus spp. were used for defining the susceptibility and resistance
Detection of plasmids
Plasmid sequences were found in 20% (6/30) of the L. monocytogenes isolates. Three different types of plasmids — LMIVRS16815, pS86, pEF1071 — were identified across six L. monocytogenes genomes isolated from pre- and post- graze fecal samples collected in 2022. Among these, five isolates harbored LMIVRS16815, three carried pS86, and one isolate (BCW14363) contained all three plasmids.
Discussion
Over the four-year (2019—2022) ICLS field trial conducted in CA and MN, we observed a higher number of L. monocytogenes positive cases in goat fecal samples in MN compared to those from sheep in CA [28, 29]. None of the grazing animals showed clinical symptoms such as neurological signs. One soil isolate collected from MN was genetically identical to goat fecal isolates collected in the same year based on whole genome comparisons. This finding suggests potential contamination of L. monocytogenes from feces to soil, with possible spread to an adjacent block via wind or other environmental vectors, as the isolate was detected in a non-grazed adjacent block. However, given that the proportion of L. monocytogenes-positive soil samples collected from MN was 0.2% (1/540), and that this soil isolate was detected only at 7 days post grazing and was not afterwards over 143 days post grazing, our results indicate that L. monocytogenes did not persist in this agricultural environment after contamination events occurred. [29].
We further characterized the genetic diversity, lineage classification, and potential genetic risk profiles of the isolates based on clonal relationships, virulence genes, and antimicrobial resistance genes. Except for the two isolates, one from sheep (lineage II) and one from goats (lineage III), all other 28 isolates in this study belonged to lineage I. This finding contrasts with the established understanding of L. monocytogenes evolutionary lineages: isolates from natural environmental sources or food products (e.g., ready-to-eat foods, dairy products) have been associated with lineage II, while lineage I strains are more commonly linked to severe clinical listeriosis cases in both humans and ruminants [16, 18, 64]. Similarly, studies conducted in Europe by Dreyer et al. (2016) and Papić et al. (2019) reported that L. monocytogenes isolates from natural environmental sources, such as feed, manure, soil and healthy ruminant feces, were significantly associated with lineage II, whereas isolates from ruminant clinical cases of rhombencephalitis or neurolisteriosis were predominantly associated with lineage I [20, 23]. In contrast, studies from the US reported that both lineage I and II isolates were commonly identified from neurologic and bacteremia cases in ruminants [22], and that lineage II was the most frequently associated with ruminant listeriosis with clinical manifestation [21]. Nevertheless, our results indicate that lineage I is not restricted to clinical cases but can also be present in healthy small ruminants and their surrounding environmental samples.
Regarding the clonal complexes (CCs) of the goat fecal isolates in this study, isolates were identified as CC1 (one isolate), CC4 (nine isolates), or CC554 (19 isolates). Similarly, Papić et al. (2019) found that CC1 and CC4 were the most prevalent CCs among isolates from both animal clinical cases and natural environmental sources [20]. Palacios-Gorba (2021) also reported that CC1 and CC4 were the most frequent CCs in fecal samples from ruminants, including cattle, sheep, and goats [19]. In a study analyzing the distribution of CCs among serotype 4b L. monocytogenes isolates from North America, CC4 was significantly overrepresented in human clinical isolates compared to food and other sources [65]. Furthermore, in a study integrating human epidemiological and clinical data with bacterial population genomics, both CC1 and CC4 were recognized as hypervirulent CCs and strongly associated with human clinical infections [66]. Interestingly, CC554 (ST554) was identified in a listeriosis outbreak in Illinois and Michigan in 2014, linked to fresh produce (mung bean sprouts), which had not been previously reported in listeriosis outbreak at that time [10]. Therefore, considering that the majority of isolates identified in this study belong to CC554 and CC4, there is a potential for these L. monocytogenes strains to pose a public health risk if transferred to fresh produce [29]. However, none of the produce samples tested positive, and only one soil sample (7 days after grazing) was positive in this study, suggesting that limited L. monocytogenes transfer occurred from ruminant feces to the ICLS environment.
Listeria Pathogenicity Islands (LIPIs) are key gene clusters in L. monocytogenes that contribute to its virulence, as they encode essential virulence factors commonly found in pathogenic strains [12]. All isolates in this study contained the complete LIPI-1 (prfA, plcA, plcB, actA, mpl, and hly genes), which is involved in the intracellular infection cycle, although a few isolates lacked the actA gene. LIPI-3, encoding a listeriolysin S, was absent from a sheep fecal sample (lineage II), but was detected in all goat fecal isolates, which were all classified as lineage I. This observation is consistent with previous reports that LIPI-3 is exclusively associated with lineage I [67]. The widespread presence of LIPI-1 and restricted distribution of LIPI-3 among lineage I strains have also been observed in earlier genomic studies of L. monocytogenes from ruminants and ready-to-eat foods in the US [21, 68]. These findings confirm that the isolates derived from these asymptomatic grazing ruminants possessed the key gene cluster profiles generally associated with L. monocytogenes pathogenicity.
Regarding virulence-associated genes, a key distinguishing feature between isolates belonging to serogroups IVb (classified as CC1 or 4) and IVb-v1 (classified as CC554) in goat fecal samples was the differential presence of the inlJ or actA genes. The inlJ gene, a recently identified member of the internalin family related to anchoring to the cell wall, is conserved in pathogenic serovars of L. monocytogenes and serves as a virulence determinant [62, 69, 70]. Accordingly, seven isolates belonging to serogroups IVb, known to be more hypervirulent, harbored the inlJ gene in this study, while none of the isolates in the serogroup IVb-v1 contained it. In contrast, the 19 isolates classified as serogroup IVb-v1 harbored the ActA gene, which enables actin polymerization and cell-to-cell spread, thereby contributing to bacterial aggregation and biofilm formation [61]. Notably, serogroup IVb-v1 comprised the majority of goat fecal isolates in this study, and was the only serogroup consistently detected across all three years of sampling (2020–2022).
Environmental survival and persistence of L. monocytogenes are facilitated by its ability to form biofilms and the presence of stress tolerance genes such as SSI-1 [71–73]. In this study, SSI-1 was detected in only one isolate obtained from a sheep fecal sample, whereas none of the isolates from goats carried this islet gene. Keeney et al. (2018) reported that the presence of SSI-1 is serotype-dependent and associated with strong biofilm formation [74]. Specifically, serotype 1/2b (serogroup IIb) predominantly harbored SSI-1 and formed strong biofilms, while serotype 4b (serogroups IVb or IVb-v1) typically lacked SSI-1 and showed weak biofilm formation. This serotype-specific distribution may explain the absence of SSI-1 among the goat isolates in this study, which were mostly classified as serogroups IVb or IVb-v1. Instead, we found that all isolates, except one, contained the agrA genes. The agr communication system regulates adhesion and biofilm formation and is essential for optimal survival of L. monocytogenes in soil [75]. However, most isolates (29/30) also harbored luxS, which is known to be associated with the inhibition of attachment and biofilm development [76].
The antibiotic resistance genes identified from this study (i.e., fosX, mprF, Lin, lmo0919, norB, and MDrL) and phenotypic antimicrobial susceptibility testing results showed consistent patterns across all 30 isolates. All isolates demonstrated resistance against fosfomycin (fosX), lincomycin (Lin), and fluoroquinolone (norB), which aligns with findings from other studies investigating L. monocytogenes isolates from ruminants, fresh produce, or wildlife samples in the US [21, 77, 78]. The presence of the fosX gene across all isolates was expected, as L. monocytogenes is known to be naturally resistant to Fosfomycin [79]. All isolates harbored the Lin gene and showed intermediate susceptibility or resistance to clindamycin in phenotypic testing, both of which are associated with the lincosamide class. Notably, none of the isolates exhibited resistance to penicillin or ampicillin, in agreement with previous studies [78, 80, 81]. Given that penicillin or ampicillin alone or in combination with gentamicin is recommended as the first-line therapy for human listeriosis [82], the observed susceptibility of all isolates to these antibiotics is clinically favorable. This is also significant from a One Health standpoint, demonstrating that these livestock-derived L. monocytogenes isolates retained susceptibility to antimicrobial agents used to treat both human and ruminant listeriosis cases (e.g., penicillin, ampicillin and oxytetracycline).
This study has some limitations. Fecal samples were collected from the field immediately after grazing events, across different blocks of the field, but within the same flock. Therefore, it is possible that some L. monocytogenes isolates originated from the same individual animal, which may have contributed to the high genomic similarity among them. Additionally, only one L. monocytogenes isolate was recovered from sheep fecal samples over the three-year period. This limited our ability to conduct meaningful genomic comparisons between sheep and goat isolates. Phenotypic antimicrobial susceptibility testing was performed using veterinary antimicrobial panels, which did not include several antibiotics widely used in human medicine (e.g., ciprofloxacin, tetracycline). Furthermore, standardized MIC breakpoints for veterinary antibiotics in L. monocytogenes are not well-established, complicating the interpretation of resistance results. Lastly, although unlikely, some genes may appear to be missing in our genome analysis due to annotation differences between our data and the reference databases.
Conclusions
By conducting WGS of L. monocytogenes isolates obtained from the ICLS field trials, genomic characterization indicated that goat fecal isolates may pose a potential public health risk if transferred to fresh produce grown in ICLS fields. Most of the isolates belonged to CC1 and CC554, both of which have been relevant to human listeriosis outbreaks. The isolates also harbored virulence associated genes, including LIPI-1 or LIPI-3 genes related to intracellular infection and survival within hosts, and agrA genes associated with biofilm formation and enhanced persistence in the agricultural soil. However, all isolates were susceptible to penicillin, ampicillin, and gentamicin, suggesting that they remain treatable with commonly used human and ruminant antimicrobial agents, an important consideration when reviewing the results from a One Health perspective. Since one soil isolate was genetically identical to goat fecal isolates collected in the same year based on whole genome comparisons, which implies that fecal isolates from asymptomatic goats could be potentially transferred to soil and survive up to seven days post grazing. Although we could not find enough isolates from sheep feces due to the practical constraints of grazing implementation in real world ICLS settings, further genomic investigation of L. monocytogenes isolates from asymptomatic sheep is needed.
Supplementary Information
Supplementary Material 1.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Cartwright EJ, Jackson KA, Johnson SD, Graves LM, Silk BJ, Mahon BE. Listeriosis outbreaks and associated food vehicles, United States, 1998–2008. Emerg Infect Dis. 2012;:1–9. 10.3201/eid 1901.10.3201/eid 1901.120393 PMC 355798023260661 · doi ↗ · pubmed ↗
- 2Van Walle I, Björkman JT, Cormican M, Dallman T, Mossong J, Moura A, et al. Retrospective validation of whole genome sequencing-enhanced surveillance of listeriosis in Europe, 2010 to 2015. Eurosurveillance. 2018;23. 10.2807/1560-7917.ES.2018.23.33.1700798.10.2807/1560-7917.ES.2018.23.33.1700798 PMC 620525330131096 · doi ↗ · pubmed ↗
- 3Cooley MB, Quinones B, Oryang D, Mandrell RE, Gorski L. Prevalence of shiga toxin producing Escherichia coli, Salmonella enterica, and Listeria monocytogenes at public access watershed sites in a California Central Coast agricultural region. Front Cell Infect Microbiol. 2014;4. 10.3389/fcimb.2014.00030.10.3389/fcimb.2014.00030 PMC 394096624624367 · doi ↗ · pubmed ↗
- 4Andrews S. A quality control tool for high throughput sequence data. 2023. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- 5Seemann T. Shovill: Assemble bacterial isolate genomes from Illumina paired-end reads. 2017. https://github.com/tseemann/shovill.
- 6Seemann T. mlst: Scan contig files against traditional Pub MLST typing schemes. 2015. https://github.com/tseemann/mlst.
- 7Seemann T. AB Ricate: Mass screening of contigs for antimicrobial resistance or virulence genes. 2015. https://github.com/tseemann/abricate.
- 8Bonin N, Doster E, Worley H, Pinnell LJ, Bravo JE, Ferm P, et al. MEGA Res and AMR++, v 3.0: an updated comprehensive database of antimicrobial resistance determinants and an improved software pipeline for classification using high-throughput sequencing. Nucleic Acids Research. 2023;51:D 744–52. 10.1093/nar/gkac 1047.10.1093/nar/gkac 1047 PMC 982543336382407 · doi ↗ · pubmed ↗