PTPN11-Related Noonan Syndrome Predisposes to Multifocal Low-Grade CNS Tumors Harboring FGFR1 Variants
Gary Kohanbash, Scott Ryall, Sam E. Gary, Lindsey M. Hoffman, Robert Siddaway, Anne E. Bendel, Karen W. Gripp, Andrew W. Walter, Jordan R. Hansford, Amy A. Smith, Hong Wang, John M. Skaugen, Uri Tabori, Cynthia E. Hawkins, Alberto Broniscer

TL;DR
People with Noonan syndrome caused by PTPN11 mutations are more likely to develop brain tumors, which often involve FGFR1 gene changes and may benefit from new treatments targeting these genes.
Contribution
Identifies a strong link between PTPN11-related Noonan syndrome and multifocal low-grade CNS tumors with FGFR1 variants, suggesting new therapeutic strategies.
Findings
Most Noonan syndrome patients with PTPN11 mutations developed CNS tumors like low-grade gliomas and DNETs.
FGFR1 abnormalities were found in 93% of tumors from Noonan syndrome patients with PTPN11 mutations.
Two specific PTPN11 genotypes were statistically associated with brain tumor occurrence in Noonan syndrome.
Abstract
To characterize the clinical, radiological, and molecular characteristics of CNS tumors associated with Noonan syndrome (NS) and other non-Neurofibromatosis type 1 RASopathies. Twenty-four patients with concern for NS underwent clinical and central radiological review in this multi-institutional study. Whole-exome sequencing, RNA sequencing, and methylation analyses of peripheral blood and/or tumor specimens were performed. Nineteen (79%) of 24 participants had NS, 17/19 (89%) of which had a germline PTPN11 variant; Nineteen of 24 participants (79%) were male. Seventeen (89%) patients with NS developed CNS cancers, including low-grade glioma, (LGG; pure pilocytic/pilomyxoid astrocytoma; n=9) and mixed dysembryoplastic neuroepithelial tumor (DNET; n=6). Five patients incidentally diagnosed did not undergo histological confirmation. Radiological review showed multifocal parenchymal…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
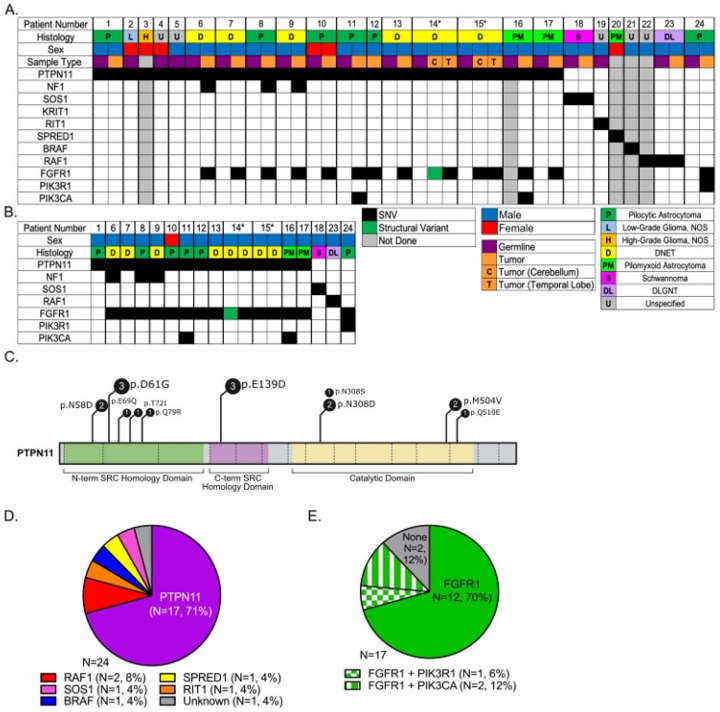 Figure 1
Figure 1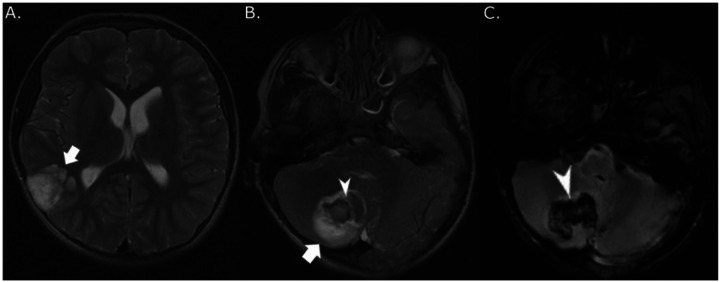 Figure 2
Figure 2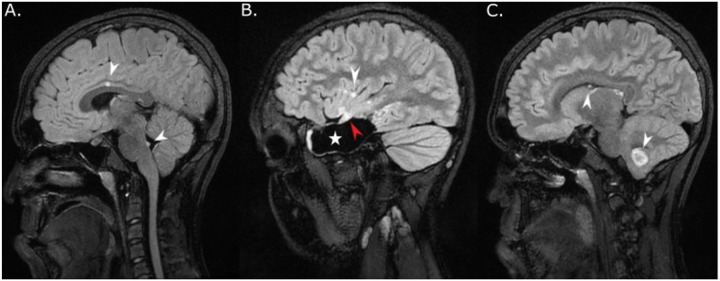 Figure 3
Figure 3Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsProtein Tyrosine Phosphatases · Neurofibromatosis and Schwannoma Cases · Fibroblast Growth Factor Research
Introduction
Cancer predisposition syndromes are present in 8% to 21% of pediatric patients with central nervous system (CNS) cancers [1–3]. Inhibition of RAS signaling through germline abnormalities in NF1, as well as germline abnormalities in non-NF1 RASopathies (NNFRAS), is associated with non-CNS cancers [4–6]. NNFRAS comprise a large and heterogenous group of disorders, the most common of which is Noonan syndrome (NS), with an incidence of up to 1:1,000 live births [7, 8]. Multiple case reports and one case series (n = 5 patients) have described the association of NNFRAS [5, 9–20], particularly NS, with low-grade glioma (LGG). However, no large-scale data exist that that characterize clinical, radiological, and molecular features driving tumorigenesis and patient outcomes [11, 14, 20, 21].
Based on the prevalence of NS, the overlap between germline abnormalities found in NNFRAS and somatic changes found in pediatric CNS cancers, and our institutional experience [7, 8, 22], we conducted this multi-institutional study consisting of the largest reported patient population to evaluate clinical, radiological, and molecular characteristics that underlie NS and development of LGG. Leveraging this large cohort, we unveil novel, consistent associations between NS and LGG, including multifocal involvement and co-occurrence of germline PTPN11 and somatic FGFR1 abnormalities, indicating potentially new FGFR1-targeted treatment options.
Material and Methods
Data and sample collection.
Once Institutional Review Board approval (University of Pittsburgh #19060202) was obtained at six participating institutions in the US and Australia, patients with NNFRAS and CNS cancers were recruited to this study. CNS cancers were either histologically confirmed or suspected based on imaging characteristics. Except for three deceased patients, informed consent to study participation was obtained from adult patients, parents, or legal guardians. Assent and re-consent at age of majority were obtained according to institutional standards. Clinical information consisting of demographics, characteristics of NNFRAS, diagnosis and therapy of CNS cancers, and survival were obtained from all research participants. An experienced pediatric neuro-oncologist (A.B.) performed central radiological review of pertinent scans, including MRI and CT at diagnosis and at most recent available follow-up.
Whole Exome (WES) and RNA Sequencing.
Germline DNA was extracted from either blood or saliva, and DNA and RNA were extracted from tumor samples, mostly derived from formalin-fixed, paraffin-embedded (FFPE) tissue, following standard methods. WES was performed at Novogene Corporation Inc. (Sacramento, CA) or at UPMC Hillman Cancer Center Core Laboratory. For WES done at Novogene, extracted DNA was quantified using Qubit TM dsDNA HS Assay Kit (Thermo Fisher Scientific, Waltham, MA). DNA libraries were prepared using Agilent SureSelect system (Agilent Technologies, Santa Clara, CA) to capture DNA coding sequences. 400 ng of genomic DNA was processed through fragmentation, enzymatic end-repair and A-tailing, adapter ligation, RNA probe hybridization, and polymerase chain reaction (PCR) amplification. The quality of libraries was analyzed with the DNA NGS 3K Assay Kit and LabChip GX Chip (PerkinElmer, Waltham, MA). Libraries with an average size of 400 bp (range: 300–600 bp) were quantified by qPCR using the VAHTS Library Quantification Kit for Illumina (Vazyme, Nanjing, China). The libraries were normalized and pooled as per the manufacturer’s protocol. Sequencing was performed using the NovaSeq 6000 platform (Illumina, San Diego, CA) with 151 paired-end reads to an average target depth of 100X for germline and tumor samples. Methods used for WES at Hillman Cancer Center were previously published [23]. Hybridization of 100 ng of FFPE-derived RNA was performed using the Illumina TruSeq exome protocol following the manufacturer’s instructions. Sequencing followed methods previously described [23].
Computational Analysis.
Raw exome sequencing reads were trimmed with Trimmomatic-v0.32 [24], aligned with bwa-mem-v0.7.8 [25], and processed with the GATK suite [26]. Somatic variants were called using the GATK HaplotypeCaller and Mutect2 [27], retaining those identified in both. Variants with coverage > 10X, a minimum variant allele frequency (VAF) > 0.05, which were predicted to be pathogenic in silico via PolyPhen2 [28], SIFT [29], and FATHMM were annotated using SnpEFF-v4.3k [30, 31]. Population filters were applied using GnomAD and variants arising in < 1% of the total population were retained [32]. Priority was assigned to the genes highlighted in Supplemental Table 1 given their known associations with RASopathies and/or gliomas. Copy number variants (CNVs) were identified using both on- and off-target reads with CNVkit v0.8.6 [33]. Raw sequencing reads were processed with the GenPipes framework [34]. Reads were qualitatively trimmed with Trimmomatic-v0.32 and aligned to human genome build GRCh37-v75 using STAR-v2.5.0 [24, 35]. Fusion transcripts were identified using MetaFusion [36]. Results were filtered against an in-house database of fusions known to be clinically relevant and consisting of a minimum of three supporting split reads. Internal tandem duplications in FGFR1 were identified using CICERO [37].
DNA Methylation Profiling.
DNA whole-genome bisulfite conversion was performed using the EZ DNA Methylation kit (Zymo Research). DNA from FFPE tissue was subsequently restored by using the Infinium FFPE DNA Restore kit (Illumina). Bisulfite-converted DNA was amplified, fragmented, and purified using the Infinium MethylationEPIC BeadChip Kit (Illumina) according to the manufacturer’s protocol, then hybridized to the BeadChip array (Illumina). The BeadChip array was washed, prepared, stained, and scanned on the Illumina NextSeq 550 (Illumina) per the manufacturer’s protocol. iDAT files were uploaded and classified using version 11b6 and 12.5 of the CNS tumor methylation classifier (https://www.molecularneuropathology.org/mnp/). A calibrated score cutoff of 0.9 was used to determine a successful diagnosis. Copy number variation (CNV) profiles were inferred using the R “conumee” package (http://bioconductor.org/packages/conumee/) as implemented in the classifier package.
Statistical analysis.
Analysis of correlation between genotype and occurrence of brain tumors in patients with NS was performed by comparing patients’ germline findings in the current study with previously published data using its 95% confidence interval (CI) [38].
Results
Patient Demographics and Clinical Outcomes
Twenty-four patients were enrolled in this study, 19 (79%) of which were male. Twenty-two patients were younger than 18 years when diagnosed with CNS cancer. Clinical diagnoses consisted of NS (n = 19; 79%), cardiofaciocutaneous (n = 2), Legius, linear nevus sebaceous (LNS), and NS with multiple lentigines syndrome in one patient each (Table 1). All patients had overt phenotypes associated with NNFRAS and were diagnosed before identification of CNS cancers. Five patients without symptoms attributable to CNS tumors were incidentally diagnosed with tumors during work-up for initiation of growth hormone supplementation (n = 2), seizures (n = 2, both with infra-tentorial tumors), or developmental delay (n = 1). After a median follow-up of 7.5 years (range: 0.5–20.2+), one patient with a low-grade glioma died of tumor progression and 3 patients died of causes unrelated to tumor. Ten patients with LGGs received chemotherapy and/or targeted therapy and one with a high-grade glioma underwent local radiation therapy and chemotherapy. None of the patients with histologically confirmed or presumptive DNETs required anticancer therapy (Table 1).
Germline Genetic Characteristics
Molecular germline analysis via WES of peripheral blood confirmed the diagnoses in 23 (96%) cases (Fig. 1A) and was used to compare to WES performed from tumor tissue (Fig. 1B). Germline pathogenic, gain-of-function PTPN11 missense variants occurred in 17 (89%; 95% CI: 0.67–0.99) patients with NS (Table 1, Fig. 1A). The distribution of germline variants in PTPN11 is shown in Fig. 1C. Two PTPN11 genotypes, NM_002834.5(PTPN11):c.182A > G (p.Asp61Gly) and c.417G > T (p.Glu139Asp) were significantly overrepresented in our patients at a two-sided 0.05 significance level (Supplemental Table 2). The second most common NNFRAS driver mutation was RAF1 (n = 2, [Figure 1D]). Interestingly, 12 (71%) patients with NS had germline FGFR1 genetic abnormalities, including two with FGFR1 and PIK3CA abnormalities and one with FGFR1 and PIK3R1 abnormalities (Fig. 1E).
Genetic and Transcriptomic Characterization of Tumor Specimens
To better understand the genetic alterations associated with NS and CNS cancers, we performed WES from FFPE tumor specimens. WES was completed in 15 tumors from 13 (81%) of 16 patients with PTPN11-associated NS and a confirmed or suspected LGG, including paired multifocal tumors in 2 patients. Fourteen (93%) of 15 tumors harbored a somatic heterozygous FGFR1 hotspot mutation or an internal tandem duplication (ITD) of its tyrosine kinase domain (n = 1) (Fig. 1B). While the cerebellar DNET of patient 14 harbored an FGFR1 ITD, his temporal DNET had an NM_023110.3(FGFR1):c.1966A > G (p.Lys656Glu). Patient 15 had an NM_023110.3(FGFR1):c.1638C > A (p.Asn546Lys) in his cerebellar and temporal DNETs, respectively.
To identify splicing events and other alterations at the transcript level, RNA sequencing was performed on 12 tumors collected from 10 patients. An FGFR1 ITD was detected in one of patient 14’s tumors. No other transcript fusions were detected.
Epigenetic Characterization of Tumor Specimens
Whole-genome bisulfite sequencing was performed on 15 tumors, leading to changes in diagnosis for two patients. Patient 3 likely had a diffuse midline glioma, H3 K27M-mutant with CDK4 and MDM2 amplification (calibrated score of 0.87). Despite histological diagnosis of pilocytic astrocytoma, DNA methylation clustered patient 24’s tumor with rosette-forming glioneuronal tumors (calibrated score of 0.99). A summary of DNA methylation results is provided in Supplemental Table 3.
Central Radiological Review and Histological Characteristics
Detailed radiological data for all cases are provided in Supplemental Table 4. Primary tumor site was in the infra-tentorial space (n = 9), cerebral cortex (n = 8), diencephalon (n = 3), optic pathway (n = 2), and spinal cord (n = 1). Patient 14 had two separate synchronous tumors of equal size in the temporal lobe and cerebellum at diagnosis (Fig. 2) [16]. Nineteen patients underwent histological tumor confirmation, and five of six asymptomatic cases were diagnosed based on imaging only.
Twelve (52%) of 23 evaluable patients, including 9 (47%) with NS, displayed multiple T_2_/fluid-attenuated inversion recovery (FLAIR) hyperintense, non-infiltrative, mostly non-enhancing and cystic, separate lesions deep in brain parenchyma without mass effect (Fig. 2 and Supplemental Table 4). Institutional radiological review had already prospectively detected multiple brain lesions in at least 9 cases. Two patients with NS (patients 14 and 15) underwent resection of two separate synchronous and metachronous tumors, respectively, in the temporal lobe and cerebellum (Supplemental Table 4). Among 10 patients without histological confirmation of distant parenchymatous lesions, a multifocal neoplastic process was confirmed based on slow radiological progression over a median of 6.6 years (range, 2.5 + to 20.2+) in 8 patients with NNFRAS, 6 of them with NS (Supplemental Table 4). Two patients had non-progressive multifocal distant tumors suspicious for a neoplastic process since they either shared imaging characteristics with the primary site (FLAIR ring sign [patient 9, Fig. 3]) or displayed mass effect with elevated choline and decreased N-acetylaspartate (NAA) on MR spectroscopy (patient 21)[39]. Multifocal brain lesions affected the posterior fossa (cerebellum and/or brainstem) and thalamus in 9 (100%) and 5 (56%) patients with NS, respectively, which are unusual locations considering their confirmed (n = 5) or presumptive (n = 2) diagnosis of dysembryoplastic neuroepithelial tumor (DNET). Five patients experienced intra-tumoral hemorrhage with or without intra-ventricular spread before diagnosis, including both patients with multifocal, histologically confirmed tumors (Supplemental Table 4). Excluding one participant with a diffuse leptomeningeal glioneuronal tumor (DLGNT), which almost uniformly presents with metastatic disease [10], five patients developed leptomeningeal tumor spread at follow-up confirmed radiologically only, including patient 9 who had a DNET and slowly progressive arachnoid disease (Fig. 3).
According to institutional histological review, diagnoses were pilocytic/pilomyxoid astrocytoma (WHO grade 1; n = 9 [47%]), DNET (WHO grade 1; n = 6 [32%]), and one each of LGG not otherwise specified (NOS; WHO grade 1), high-grade glioma NOS, schwannoma (WHO grade 1), and DLGNT (Fig. 1A). DNETs were diagnosed in both patients with histologically confirmed multifocal tumors. Central histological review of these 19 cases was not performed.
Discussion
Our work provides the first large-scale clinical, radiological, and molecular study evaluating NS-associated brain neoplasms, which were mostly LGGs. There was an overwhelming male predominance observed in NS (79%), similar to previously published case reports (61%; Supplemental Table 5) [5, 9–20], for which this sex bias remains unknown. Brain tumors developed during childhood in most patients with NS and their continued slow growth during adulthood was documented in several cases. Further work is warranted to determine the behavior and repercussions of NS-associated brain tumors in adults.
The prevalence of germline PTPN11 variants in NS is approximately 45% [40]. Herein, we demonstrated an overrepresentation of germline PTPN11 and a genotype-phenotype association between NM_002834.5(PTPN11):c.182A > G (p.Asp61Gly) or c.417G > T (p.Glu139Asp) and the development of brain tumors in patients with NS. These PTPN11 variants, as well as others, are known to induce protein changes and activate mitogen-activated pathway kinase (MAPK) signaling [38]. Additionally, we discovered a 93% co-occurrence rate of somatic FGFR1 abnormalities and PTPN11 germline variants in patients with NS and LGG. A previously reported in vitro cooperative effect of abnormalities in both genes was shown to cause overexpression of pERK compared to PTPN11 variants alone in murine fibroblasts and was implicated in the tumorigenesis of pilocytic astrocytomas [41]. Similarly, a recent analysis of FGFR alterations across gliomas, including pediatric gliomas, support the hypothesis of a synergistic effect of PTPN11 and FGFR1 in the genesis of gliomas [42]. We propose that cooperative interaction between PTPN11 and FGFR1 abnormalities is critical for the tumorigenesis of LGGs in patients with NS, including DNETs. This has potential practical implications for patients with NS, as FGFR1 inhibitors have shown preliminary activity in LGGs harboring FGFR1 variants [43]. Likewise, other inhibitors of the MAPK pathway (e.g., MEK inhibitors) may represent alternative therapies for patients with NS and LGG.
Multiple case reports described the presence of separate parenchymal lesions in patients with NS and NNFRAS [9, 17, 19], but only a few patients underwent histological confirmation [16, 44, 45]. Here, we show that multifocal parenchymal lesions in NS unequivocally represent synchronous or metachronous LGGs based on histological confirmation (n = 2; 22%), slow growth documented over a median follow-up of 6.6 years (n = 6; 67%), or typical radiological characteristics (n = 1, 11%). The only patient with NS without histological confirmation or radiological disease progression harbored a synchronous satellite lesion located in the cerebellum with FLAIR hyperintense ring sign, similar to the primary tumor, which is suggestive of DNETs [39]. Although this case was previously reported, no details about radiological findings had been provided [46]. The prevalence of multifocal tumors in 9 (53%) of 17 patients with NS and LGGs, a rate at least twice higher than those for patients with NF1 based on imaging [47], is striking and warrants confirmation in larger studies. Unlike NF1, only two (11%) of our patients with NS had primary involvement of the optic pathway [47], which further corroborates the distinct biological, clinical, and radiological tumor characteristics in patients with NS.
DNETs and other low-grade glioneuronal tumors accounted for approximately 50% of all CNS tumors in patients with NS and other NNFRAS reported to date [5, 10, 13, 14, 17–20, 45, 46, 48] (Supplemental Table 5). Likewise, DNETs were overrepresented and commonly displayed multifocal involvement in our patients, an otherwise rare phenomenon [49]. Therefore, the diagnosis of NS or other NNFRAS should be investigated in patients suspected to harbor multifocal LGGs. Based on a previous report [44], it is possible that individuals with NS and other NNFRAS could have histologically distinct multifocal LGGs. We also report a few unique cases, including the first association of Legius syndrome with a CNS cancer and a second case of a presumed brain tumor in a child with cardiofaciocutaneous syndrome. Unfortunately, no tumor tissue was available in either patient for molecular analysis.
Although most patients did not require anticancer therapy beyond surgery, 7 (41%) of 17 patients with NS and a confirmed or presumptive LGG required standard chemotherapy and/or targeted agents, 5 of whom %) received at least 2 different regimens. Overall, NS-associated CNS tumors are mostly non-life-threatening LGGs with indolent growth, but may present with complications, including disease progression, leptomeningeal spread, significant morbidity, and even death despite aggressive therapy.
One limitation of this study is its mostly retrospective design. Although 7 patients had been included in other reports [9–11, 16, 45, 46], we generated extensive new clinical, radiological, and molecular data about them. Based on the patients’ overt presentation of NS and NNFRAS, our findings may not be applicable to those with subtle features who are diagnosed based on molecular screening after the diagnosis of CNS tumors [17].
In conclusion, patients with NS and germline PTPN11 variants account for nearly all individuals with NNFRAS and brain tumors in this study. Although approximately half of these patients harbored multifocal LGGs, their prognosis was good. However, anticancer therapy is required in a substantial minority of patients, particularly those with DNETs. Our finding that 93% of patients with NS and LGG harbor FGFR1 mutations provides an exciting new therapeutic opportunity for these patients. Further investigation with CNS imaging is recommended for patients with NNFRAS presenting with neurologic signs and/or symptoms. Screening of asymptomatic patients with NM_002834.5(PTPN11):c.182A > G (p.Asp61Gly) or c.417G > T (p.Glu139Asp) variants for brain tumors may also be considered.
Supplementary Material
Supplementary Files
This is a list of supplementary files associated with this preprint. Click to download.
SupplTable2GenotypeDistribution.xlsx
SupplTable3DNAMethylation.xlsx
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Fiala EM, Jayakumaran G, Mauguen A, Kennedy JA, Bouvier N, Kemel Y, Fleischut MH, Maio A, Salo-Mullen EE, Sheehan M, Arnold AG, Latham A, Carlo MI, Cadoo K, Murkherjee S, Slotkin EK, Trippett T, Glade Bender J, Meyers PA, Wexler L, Dela Cruz FS, Cheung NK, Basu E, Kentsis A, Ortiz M, Francis JH, Dunkel IJ, Khakoo Y, Gilheeney S, Farouk Sait S, Forlenza CJ, Sulis M, Karajannis M, Modak S, Gerstle JT, Heaton TE, Roberts S, Yang C, Jairam S, Vijai J, Topka S, Friedman DN, Stadler ZK, Robson M, Berger MF, Schultz N, Ladanyi M, O’Reilly RJ, Abrams · doi ↗ · pubmed ↗
- 2Wong M, Mayoh C, Lau LMS, Khuong-Quang DA, Pinese M, Kumar A, Barahona P, Wilkie EE, Sullivan P, Bowen-James R, Syed M, Martincorena I, Abascal F, Sherstyuk A, Bolanos NA, Baber J, Priestley P, Dolman MEM, Fleuren EDG, Gauthier ME, Mould EVA, Gayevskiy V, Gifford AJ, Grebert-Wade D, Strong PA, Manouvrier E, Warby M, Thomas DM, Kirk J, Tucker K, O’Brien T, Alvaro F, Mc Cowage GB, Dalla-Pozza L, Gottardo NG, Tapp H, Wood P, Khaw SL, Hansford JR, Moore AS, Norris MD, Trahair TN, Lock RB, Tyrrell V, Haber M, Marshall GM, Ziegler DS, Ekert PG, Cowl · doi ↗ · pubmed ↗
- 3Zhang J, Walsh MF, Wu G, Edmonson MN, Gruber TA, Easton J, Hedges D, Ma X, Zhou X, Yergeau DA, Wilkinson MR, Vadodaria B, Chen X, Mc Gee RB, Hines-Dowell S, Nuccio R, Quinn E, Shurtleff SA, Rusch M, Patel A, Becksfort JB, Wang S, Weaver MS, Ding L, Mardis ER, Wilson RK, Gajjar A, Ellison DW, Pappo AS, Pui CH, Nichols KE, Downing JR (2015) Germline Mutations in Predisposition Genes in Pediatric Cancer. N Engl J Med 373: 2336–2346 doi:10.1056/NEJ Moa 150805426580448 PMC 4734119 · doi ↗ · pubmed ↗
- 4Kratz CP, Rapisuwon S, Reed H, Hasle H, Rosenberg PS (2011) Cancer in Noonan, Costello, cardiofaciocutaneous and LEOPARD syndromes. Am J Med Genet C Semin Med Genet 157C: 83–89 doi:10.1002/ajmg.c.3030021500339 PMC 3086183 · doi ↗ · pubmed ↗
- 5Kratz CP, Franke L, Peters H, Kohlschmidt N, Kazmierczak B, Finckh U, Bier A, Eichhorn B, Blank C, Kraus C, Kohlhase J, Pauli S, Wildhardt G, Kutsche K, Auber B, Christmann A, Bachmann N, Mitter D, Cremer FW, Mayer K, Daumer-Haas C, Nevinny-Stickel-Hinzpeter C, Oeffner F, Schluter G, Gencik M, Uberlacker B, Lissewski C, Schanze I, Greene MH, Spix C, Zenker M (2015) Cancer spectrum and frequency among children with Noonan, Costello, and cardio-facio-cutaneous syndromes. Br J Cancer 112: 1392–1397 doi:10.1038/bjc.2015.752574247 · doi ↗ · pubmed ↗
- 6Lin AL, Gutmann DH (2013) Advances in the treatment of neurofibromatosis-associated tumours. Nat Rev Clin Oncol 10: 616–624 doi:10.1038/nrclinonc.2013.14423939548 · doi ↗ · pubmed ↗
- 7Dunnett-Kane V, Burkitt-Wright E, Blackhall FH, Malliri A, Evans DG, Lindsay CR (2020) Germline and sporadic cancers driven by the RAS pathway: parallels and contrasts. Ann Oncol 31: 873–883 doi:10.1016/j.annonc.2020.03.29132240795 PMC 7322396 · doi ↗ · pubmed ↗
- 8Roberts AE, Allanson JE, Tartaglia M, Gelb BD (2013) Noonan syndrome. Lancet 381: 333–342 doi:10.1016/S 0140-6736(12)61023-X 23312968 PMC 4267483 · doi ↗ · pubmed ↗