Silane Redistribution Catalyzed by [Mes-B-TMP]+ Borinium Ion
Min-Hsiung Wu, Yu-Jiang Lin, Bo-An Chen, Ching-Wen Chiu

TL;DR
A borinium ion effectively catalyzes silane redistribution and cross-coupling reactions under mild conditions.
Contribution
The arylamino borinium ion [1]+ enables selective aryl transfer and cross-coupling by modulating Lewis acidity.
Findings
The [1]+ ion catalyzes ArMe2SiH to produce Ar2SiMe2 and Me2SiH2 under mild conditions.
The amino substituent in [1]+ prevents catalyst decomposition by modulating Lewis acidity.
The reaction extends to cross-coupling between ArMe2SiH and Et2SiH2/Et3SiH.
Abstract
Borinium ions, featuring two empty p orbitals at boron, exhibit exceptional Lewis acidity but often suffer from excessive reactivity that limits their catalytic utility. In this study, we demonstrate that the arylamino borinium ion [1]+ effectively mediates aryl transfer in ArMe2SiH to afford Ar2SiMe2 and Me2SiH2 under mild conditions. The reaction can also be extended to cross-coupling between ArMe2SiH and Et2SiH2/Et3SiH. Computational studies revealed that the amino substituent in [1]+ plays a pivotal role in modulating the Lewis acidity of the boron center, enabling reversible Si–H activation while preventing irreversible B–C bond cleavage and catalyst decomposition. These findings highlight the potential of electronically tuned borinium ions as catalysts for activating inert substrates in organic transformations.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
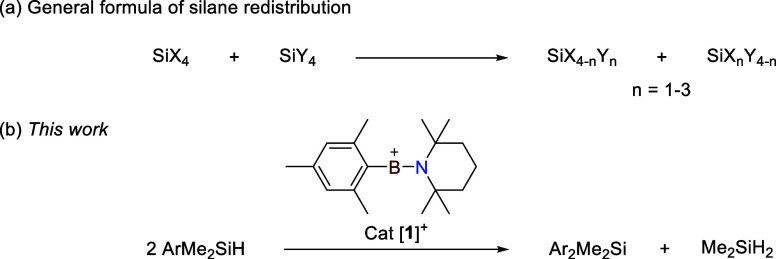 Figure 1
Figure 1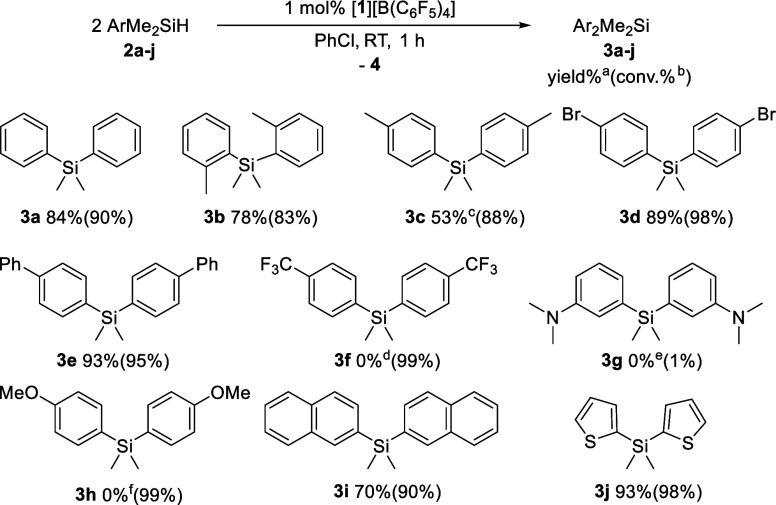 Figure 2
Figure 2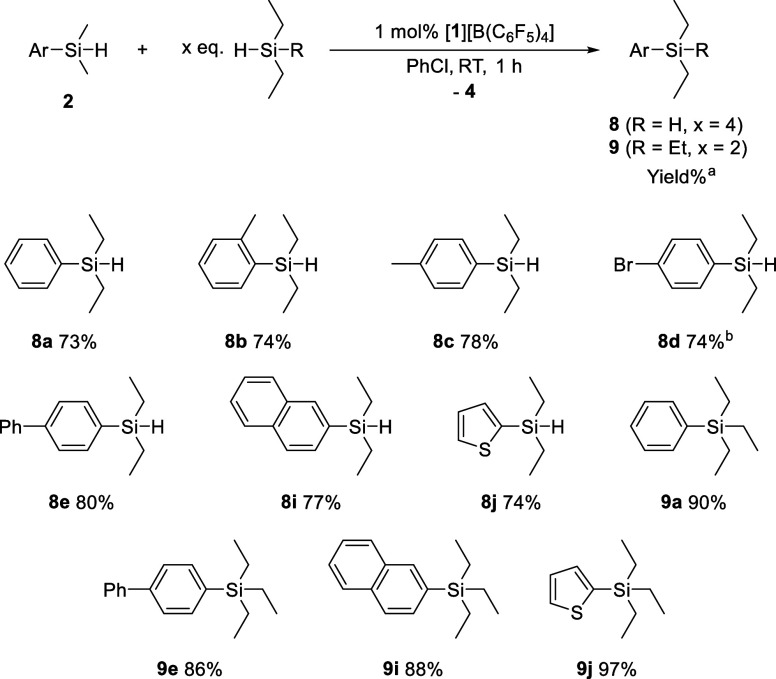 Figure 3
Figure 3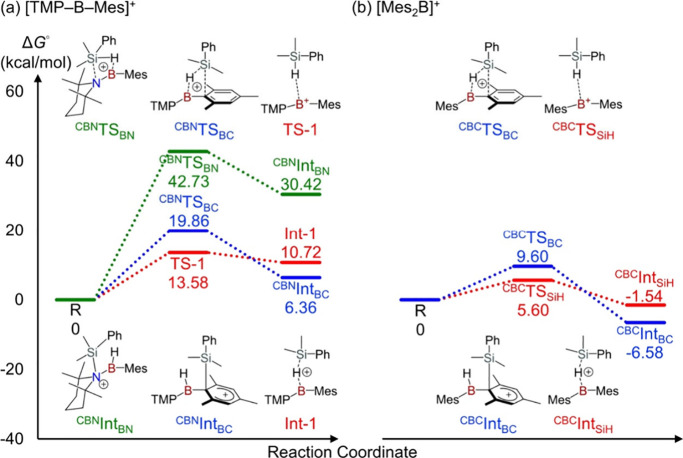 Figure 4
Figure 4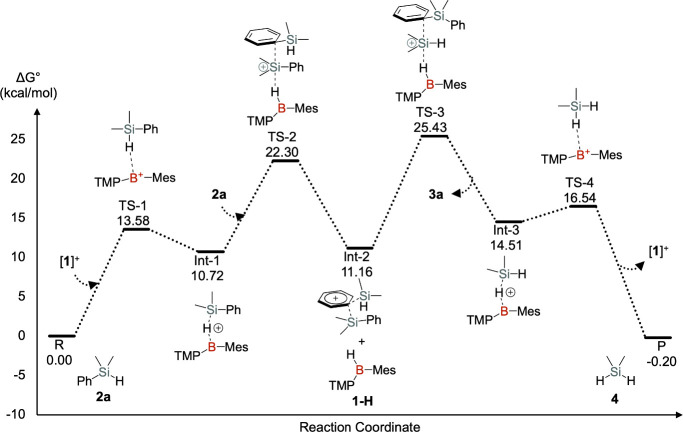 Figure 5
Figure 5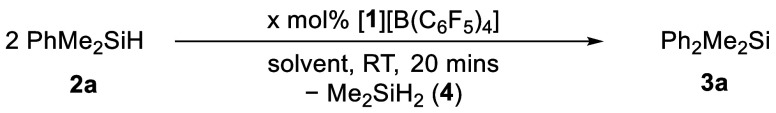 Figure 6
Figure 6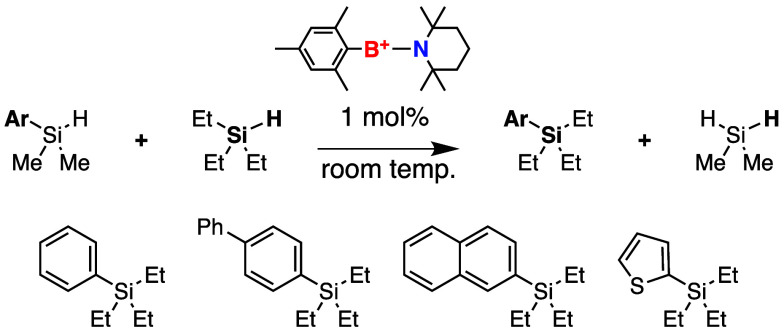 Figure 7
Figure 7- —National Taiwan University10.13039/501100006477
- —National Science and Technology Council10.13039/501100020950
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsOrganoboron and organosilicon chemistry · Catalytic Cross-Coupling Reactions · Catalytic C–H Functionalization Methods
Silanes are indispensable reagents in organic synthesis, material science, polymer chemistry, and semiconductor manufacturing. ?−? ? ? While organometallic reagents are commonly employed for the construction of Si–C bonds, silane redistribution, a process involving the exchange of substituents between silicon centers, offers an atom-economical route to generate diverse organosilanes (Figurea). ?,? Catalytic silane redistribution was first documented in the mid-19th century, when Friedel and Ladenburg observed the disproportionation of triethoxysilane (SiH(OEt)3) to silane (SiH_4_) and tetraethoxysilane (Si(OEt)4) in the presence of sodium, a process likely mediated by the ethoxide anion.? Since then, silane redistribution utilizing alkaline catalysts has been investigated in detail. ?−? ? Later, acidic metal halides were found to catalyze the redistribution of tetraalkylsilanes at elevated temperature with a catalytic order of Al_2_X_6_ > Ga_2_X_6_ > FeX_3_ ≈ BX_3_, which is consistent with their Lewis acidity as observed in electrophilic aromatic substitution reactions.? In addition, transition-metal complexes have been shown to promote silane redistribution. ?,?−? ? ? However, high reaction temperatures are generally required for these systems. In 2021, our group reported the synthesis of an arylamino borinium ion, [Mes-B-TMP][B(C_6_F_5_)4] ([1][B(C_6_F_5_)4]), and demonstrated its catalytic activity in hydrosilylation of ketones and aldehydes. ?,? We proposed that the key activation step involves the interaction of [1]^+^ with the Si–H bond of silane rather than with the carbonyl compounds. This finding prompted us to explore the potential of [1]^+^ as a Lewis acid catalyst for silane redistribution at ambient temperature (Figureb).
In 2017, Hou and co-workers demonstrated that B(C_6_F_5_)3 (BCF), a prototypical strong boron Lewis acid, can catalyze the C–Si/Si–H cross-metathesis of electron-rich hydrosilanes.? This method, however, required harsh conditions (100 °C for 24 h) and was limited to substrates possessing an m-aminoaryl group. Unfunctionalized substrates, such as PhMe_2_SiH (2a), remained intact even after heating at 100 °C for 24 h. In stark contrast, simply treating a CDCl_3_ solution of 2a with 10 mol% of [1][B(C_6_F_5_)4] at room temperature in a sealed J-Young tube rapidly afforded the redistribution product, Ph_2_Me_2_Si (3a) and Me_2_SiH_2_ (4), in 43% yield. However, the borinium ion was not effective for Ph_2_SiH_2_, Et_2_SiH_2_, or Et_3_SiH (Table S1). The reaction of Ph_2_SiH_2_ produced a complex mixture of silanes containing multiple Ph_ x SiH_4‑x _ (x = 0–4) derivatives, indicating extensive and uncontrolled redistribution. In contrast, Et_2_SiH_2 and Et_3_SiH remained largely unreacted under identical conditions, and no significant redistribution products were detected. Thus, we decided to focus on the redistribution of aryldimethylsilanes (ArMe_2_SiH).
To improve the yield and selectivity of the redistribution of 2a, we screened several solvents (Table). Despite the complete consumption of 2a in CDCl_3_ or CH_2_Cl_2_ upon the addition of [1]^+^, 3a was obtained only in moderate yields. Analysis of the reaction mixture revealed the formation of chlorosilane byproducts (e.g., PhMe_2_SiCl), suggesting the participation of the solvent in the reaction. This side reaction is consistent with the generation of silylium ion intermediates through heterolytic cleavage of the Si–H bond. ?,? To suppress chlorosilane formation, the reaction was carried out in aromatic solvents. All aromatic solvents provided comparable results, with chlorobenzene (PhCl) being marginally superior. We noticed that, despite the poor solubility of [1]^+^ in benzene, the catalytic performance remained comparable to that observed in halobenzene solvents. This observation suggested that the catalyst loading could be reduced without compromising reactivity. Indeed, a comparable yield of 3a was achieved within 20 min using only 1 mol% catalyst. Finally, both conversion and yield were further improved by conducting the reaction in an open vial inside a glovebox to continuously remove the volatile byproduct Me_2_SiH_2_ (4), thereby shifting the equilibrium toward the products (Table). ?,?
With the optimal conditions established, we next explored the substrate scope of the reaction across a series of functionalized arylsilanes (Figure). The ortho-tolyl (2b), para-bromophenyl (2d), biphenyl-4-yl (2e), 2-naphthyl (2i), and 2-thiophenyl (2j) silanes were efficiently converted to the corresponding diaryl(dimethyl)silanes in high yields. In contrast, the para-tolyl-substituted silane (2c) afforded 3c in only a moderate yield. Examination of the reaction mixture revealed the presence of a 1:1 ratio of (p-tol)3_SiMe (5) and (p-tol)SiMe_3 (6), indicating the occurrence of arene/Me exchange. Indeed, monitoring of the reaction progress by NMR spectroscopy confirmed the rapid generation of 3c in a high yield within the first 5 min (Figure S2). However, the yield of 3c subsequently declined from 80% to 50%, reaching equilibrium at 20 min with the concomitant formation of 5 and 6. The ability of [1]^+^ to activate the Si–Me bond of 3c was further confirmed by the formation of 5 and 6 (11% each) from 3c in the presence of 1 mol% of [1]^+^ (Figure S3). Unfortunately, the reaction was not applicable to substrates bearing trifluoromethyl (2f), dimethylamino (2g), or methoxy (2h) substituents. For 2g, catalyst inhibition via base coordination was observed, whereas activation of the CF_3_ group in 2f led to the formation of a fluorosilane mixture (Figures S4–S6). In the case of 2h, polymerization via a Piers–Rubinsztajn-type reaction, accompanied by methane evolution and Si–O bond formation, was observed (Figure S7). ?,?
After completing the homometathesis study of aryldimethylsilanes, we next examined whether cross-coupling reactions between two different arylsilanes could be achieved (Table S2). Treating an equimolar mixture of 2a and 2d with 1 mol% [1]^+^ afforded a mixture containing 3a, (4-Br-C_6_H_4_)PhMe_2_Si (7d), and 3d in 17%, 15%, and 1% NMR yield, respectively. Increasing the catalyst loading to 3 mol% markedly improved the yield of the cross-coupling product 7d to 54%. Attempts to enhance cross-coupling by increasing the steric bulk of the aryl group were unsuccessful. The reaction of 2a and 2b resulted in the formation of (o-tolyl)PhMe_2_Si (7b) in at most 36% yield. Notably, when 2a was reacted with the more electron-donating thiophenylsilane (2j), the cross-coupling product (2-thiophenyl)PhMe_2_Si (7j) was formed in a 74% yield. In all cases, however, substantial amounts of homocoupling products were also obtained, due to the comparable donating abilities of the aryldimethylsilane substrates. Since tuning the electron density of the aryl ring through CF_3_, OMe, or NMe_2_ substitution was not feasible, we next explored the cross-coupling reactions of aryldimethylsilanes with Et_2_SiH_2_ and Et_3_SiH.
To our delight, the cross-coupling products 8 and 9 were obtained in high yields by mixing aryldimethylsilanes with alkylsilanes in the presence of 1 mol% of [1]^+^ at room temperature (Figure). The reaction of 2a with 4 equiv of Et_2_SiH_2_ afforded the cross-coupling product 8a in 73% yield, whereas mixing 2a and 2 equiv of Et_3_SiH gave 9a in 90% yield. Because compound 8, ArEt_2_SiH, can undergo further aryl transfer reaction in the presence of a borinium ion catalyst, a higher loading of Et_2_SiH_2_ was essential to suppress secondary transformation. The reaction was successfully extended to other combinations of silanes, furnishing the corresponding cross-coupling products in high yields, including 2-tolyldiethylsilane (8b), 4-tolyldiethylsilane (8c), 4-bromophenyldiethylsilane (8d), biphenyl-4‑yldiethylsilane (8e), biphenyl-4‑yltriethylsilane (9e), 2-naphthyldiethylsilane (8i), 2-naphthyltriethylsilane (9i), 2-thiophenyldiethylsilane (8j), and 2-thiophenyltriethylsilane (9j).
To gain deeper insight into the aryl amino borinium-mediated aryl transfer process, we examined the redistribution reaction of PhMe_2_SiH computationally. Three potential reaction pathways between PhMe_2_SiH and [1]^+^ were identified, and all were found to be endergonic (Figurea). Compared to the Si–H addition across the B–N and B–C bonds, the formation of a silane-borinium σ-complex represents the most feasible initial interaction for [1]^+^ with an energy barrier of 13.58 kcal/mol. ?,? To elucidate the influence of the amino substituent at boron, we also evaluated the corresponding processes for the more Lewis acidic all-carbon-substituted borinium ion [Mes_2_B]^+^ (Figureb). ?,? Both the 1,2-addition pathway and the σ-complex formation at [Mes_2_B]^+^ were found to be exergonic with noticeably lower activation barriers, which are consistent with the higher intrinsic Lewis acidity of [Mes_2_B]^+^. However, the increased reactivity of [Mes_2_B]^+^ toward B–C bond cleavage, plausibly happening after the Si–H activation, would destroy the borinium scaffold and prevent catalytic turnover. This prediction aligns with experimental observations: while [1]^+^ serves as an efficient catalyst for silane redistribution, [Mes_2_B]^+^ decomposes upon addition of silane (Table S4).? The amine substituent in [1]^+^ plays a key role in modulating the Lewis acidity of the boron center, thereby enabling reversible Si–H activation.
A proposed reaction mechanism and the calculated potential energy surface for the [1]^+^ catalyzed redistribution of PhMe_2_SiH is shown in Figure. The initial step is the formation of a silane-borinium σ-complex (Int-1) as discussed above. The second step involves nucleophilic attack by the phenyl group of a second PhMe_2_SiH molecule on the silicon atom of Int-1, inducing heterolytic Si–H cleavage to generate the phenyl-bridged disilicon cation (Int-2) and hydridoborane (1-H).? Subsequent reverse hydride transfer from 1-H to the less sterically hindered silicon center of Int-2 yields Ph_2_SiMe_2_ and the Me_2_SiH_2_-borinium σ-complex (Int-3). The calculated energy differences among Int-1, Int-2 + 1-H, and Int-3 are small, and the highest barrier in this sequence is only 14.71 kcal/mol, indicating that these species exist in a rapid and reversible equilibrium. Therefore, removal of the volatile Me_2_SiH_2_ is essential to drive the reaction toward completion. Additionally, the calculated overall energy barrier of the reaction is 25.43 kcal/mol, which is consistent with our experimental results. The calculated reaction mechanism is comparable to the BCF-mediated aryl transfer reaction proposed by Hou’s group.?
In conclusion, we have demonstrated that the arylamino borinium ion [1]^+^ is an effective catalyst for the redistribution of unfunctionalized ArMe_2_SiH, affording Ar_2_SiMe_2_ and Me_2_SiH_2_ at ambient temperature. The aryl transfer process between silanes can be extended to diethyl- and triethylsilanes, providing the corresponding cross-coupling products in high yields. Theoretical calculations reveal that the amino substituent in the borinium ion is essential for achieving reversible Si–H activation while preventing catalyst decomposition. Overall, this study not only demonstrates the capacity of borinium ions to active inert substrates but also highlights the critical role of the borinium substituent in determining catalyst performance.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Aziz T.Ullah A.Fan H.Jamil M. I.Khan F. U.Ullah R.Iqbal M.Ali A.Ullah B.Recent Progress in Silane Coupling Agent with Its Emerging Applications J. Polym. Environ.202129113427344310.1007/s 10924-021-02142-1 · doi ↗
- 2Indumathy B.Sathiyanathan P.Prasad G.Reza M. S.Prabu A. A.Kim H.A Comprehensive Review on Processing, Development and Applications of Organofunctional Silanes and Silane-Based Hyperbranched Polymers Polymers 202315251710.3390/polym 1511251737299316 PMC 10255420 · doi ↗ · pubmed ↗
- 3Zhao Q.Geng Q.Li Y.Li J.Liu Z.Emerging applications of acylsilanes in organic synthesis and beyond Org. Chem. Front.20231051316132110.1039/D 2QO 02010 F · doi ↗
- 4Han R.Li Y.Zhu Q.Niu K.Research on the preparation and thermal stability of silicone rubber composites: A review JCOMC 2022810024910.1016/j.jcomc.2022.100249 · doi ↗
- 5Barton, T. J. ; Boudjouk, P. Organosilicon chemistry: A brief overview. In Silicon-Based Polymer Science; Zeigler, J. M. , Fearson, F. W. G. ; Advances in Chemistry 224; American Chemical Society: Washington, DC, 1989; Chapter 1, pp 3–46. 10.1021/ba-1990-0224.ch 001 · doi ↗
- 6Curtis, M. D. ; Epstein, P. S. Redistribution Reactions on Silicon Catalyzed by Transition Metal Complexes; Advances in Organometallic Chemistry 19; Stone, F. G. A. , West, R. , Eds.; Academic Press, 1981; pp 213–255.
- 7Friedel C.Ladenburg A.Ueber das Siliciumchloroform und dessen Derivate Liebigs Ann. Chem.1867143111812810.1002/jlac.18671430112 · doi ↗
- 8Beck E. W.Daudt W. H.Fletcher H. J.Hunter M. J.Barry A. J.Alkaline Rearrangement of Phenyl Groups Linked To Silicon J. Am. Chem. Soc.19598151256125710.1021/ja 01514 a 062 · doi ↗