The β-goblin gene architecture in individuals with and without sickle cell disease in Nigeria: Implications for β-thalassaemia trait diagnosis
Oluwatoyin A. Babalola, Biobele J. Brown, Foluke Fasola, Jing Zhang, Yonglan Zheng, Abayomi B. Odetunde, Adeyinka G. Falusi, Olufunmilayo Olopade

TL;DR
This study finds that β-thalassaemia trait is rare in Nigeria, even among those with sickle cell disease, and highlights the importance of genetic testing over screening methods for accurate diagnosis.
Contribution
The first molecular confirmation of β-thalassaemia trait prevalence in the Yoruba population and identification of two rare genetic variants.
Findings
Molecular analysis revealed a β-thalassaemia trait prevalence of less than 1% in both groups.
Two rare variants (rs537944366T>C and rs33915217C>A) were identified for the first time in the Yoruba population.
The β-thalassaemia mutation was found on a minor ancestral haplotype atypical of Africa.
Abstract
β-thalassaemia is considered rare in Africa; however, recent screening-based studies suggest a β-thalassaemia trait prevalence of 6% – 10% among individuals with sickle cell disease (SCD) and up to 25% in those without SCD. Co-inheritance with SCD may modify disease severity, highlighting the need for molecular confirmation. To ascertain the prevalence and genetic basis of β-thalassaemia trait in Nigerians with and without SCD. We recruited 260 participants (130 per group; aged 3 years – 69 years, median [interquartile range] = 16 [9–29]). Haemoglobin fractions were analysed using high-performance liquid chromatography, and full blood counts were obtained. A 1.6 kb region of the β-globin gene was amplified and sequenced by Sanger sequencing. Variants were annotated and haplotypes constructed. An additional 26 samples from a separate SCD cohort were also genotyped. Molecular analysis…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 FIGURE 1
FIGURE 1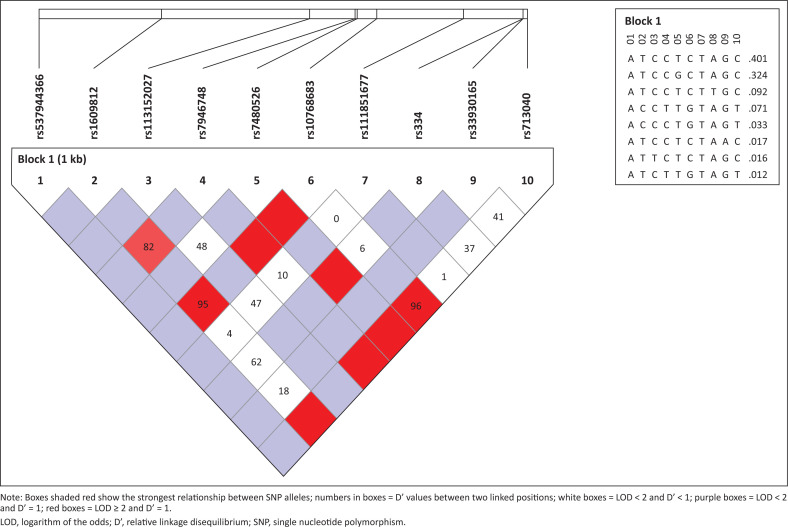 FIGURE 2
FIGURE 2| SNP rs number | HGVS nomenclature | Allele frequency in cases | Allele frequency in controls | Allele frequency in YRI | Allele frequency in Africans | Global Allele frequency | ClinVar annotation |
|---|---|---|---|---|---|---|---|
| rs713040 | 1.000 | 0.892 | 0.894 | 0.884 | 0.7140 | Synonymous mutation | |
| rs33930165 | 0.058 | 0.018 ( | 0.028 | 0.013 | 0.0030 | Missense variant | |
| rs334 | 0.942 | 0.085 ( | 0.139 | 0.100 | 0.0270 | Missense variant | |
| rs33915217 | - | - | 0.000 | 0.000 | 0.0000 | β+-thal, β-thal major | |
| rs111851677 | - | 0.014 ( | 0.019 | 0.022 | 0.0060 | β+-thal, benign (conflicting submission) | |
| rs10768683 | 1.000 | 0.892 | 0.894 | 0.890 | 0.7200 | Intron variant, benign | |
| rs7480526 | - | 0.256 | 0.338 | 0.379 | 0.3690 | Intron variant, benign | |
| rs7946748 | - | 0.119 | 0.065 | 0.065 | 0.0990 | Intron variant, benign | |
| rs113152027 | - | 0.026 | 0.009 | 0.007 | 0.0020 | Intron variant, benign | |
| rs1609812 | 1.000 | 0.889 | 0.894 | 0.884 | 0.7140 | Intron variant, benign | |
| rs537944366 | - | 0.0043 ( | - | 0.002 | 0.0004 | 3’ UTR variant, uncertain significance |
| rs numbers | rs numbers | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| rs537944366 | rs1609812 | rs113152027 | rs7946748 | rs7480526 | rs10768683 | rs111851677 | rs334 | rs33930165 | |
|
| |||||||||
| rs1609812 | 1.000 | - | - | - | - | - | - | - | - |
| rs113152027 | 1.000 | 1.000 | - | - | - | - | - | - | - |
| rs7946748 | 1.000 | 0.824 | 1.000 | - | - | - | - | - | - |
| rs7480526 | 1.000 | 1.000 | 0.480 | 1.000 | - | - | - | - | - |
| rs10768683 | 1.000 | 0.955 | 1.000 | 1.000 | 1.000 | - | - | - | - |
| rs111851677 | 1.000 | 0.043 | 0.478 | 0.102 | 1.000 | 0.000 | - | - | - |
| rs334 | 0.050 | 0.620 | 0.280 | 0.360 | 1.000 | 0.060 | 1.000 | - | - |
| rs33930165 | 1.000 | 0.180 | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 | - |
| rs713040 | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 | 0.960 | 0.011 | 0.379 | 0.412 |
|
| |||||||||
| rs1609812 | 0.001 | - | - | - | - | - | - | - | - |
| rs113152027 | 0.000 | 0.003 | - | - | - | - | - | - | - |
| rs7946748 | 0.000 | 0.508 | 0.002 | - | - | - | - | - | - |
| rs7480526 | 0.008 | 0.064 | 0.011 | 0.048 | - | - | - | - | - |
| rs10768683 | 0.001 | 0.776 | 0.004 | 0.636 | 0.076 | - | - | - | - |
| rs111851677 | 0.000 | 0.000 | 0.151 | 0.002 | 0.032 | 0.000 | - | - | - |
| rs334 | 0.040 | 0.040 | 0.050 | 0.070 | 0.370 | 0.000 | 0.050 | - | - |
| rs33930165 | 0.000 | 0.000 | 0.001 | 0.002 | 0.011 | 0.003 | 0.000 | 0.050 | |
| rs713040 | 0.000 | 1.000 | 0.004 | 0.676 | 0.073 | 0.887 | 0.000 | 0.002 | 0.001 |
| Major ancestral haplotype | Frequency | Minor ancestral haplotype | Frequency | |
|---|---|---|---|---|
| 1 | TAGGAGATCG | 0.401 | TGGAACATCA | 0.071 |
| 2 | TAGGCGATCG | 0.324 | TGGGACATCA | 0.033 |
| 3 | TAGGAGAACG | 0.092 | TAGAACATCA | 0.012 |
| 4 | TAGGAGATTG | 0.017 | - | - |
| 5 | TAAGAGATCG | 0.016 | - | - |
|
|
| - |
| |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsHemoglobinopathies and Related Disorders · Blood groups and transfusion · Genetic Associations and Epidemiology
Introduction
β-thalassaemia is primarily caused by point mutations and, less commonly, by deletions or insertions in the β-globin gene locus.^1^ This gene is highly heterogeneous, with approximately 954 documented variants, over 300 of which are associated with β-thalassaemia.^2^ β-thalassaemia major arises from the complete absence of β-globin chain production, while β-thalassaemia trait (minor) and β-thalassaemia intermedia involve a reduction in β-globin synthesis. All three forms result in decreased production of adult haemoglobin (HbA),^3,4,5^ leading to anaemia of varying severity, depending on the mutation type and zygosity.^4^
While β-thalassaemia is globally distributed, its prevalence varies. Historically, it has been endemic in the Mediterranean, the Middle East, and Southeast Asia, with the highest carrier frequencies reported in regions such as Cyprus, Sardinia, and parts of Southeast Asia.^6,7^ The global carrier rate is estimated at approximately 1.5%.^5^ However, patterns are shifting as a result of increased migration and interethnic marriages, leading to cases in previously low-prevalence areas.^5,7^
In West Africa, particularly Nigeria, β-thalassaemia was once considered rare. However, recent studies suggest a rising prevalence. In Nigeria, population-based screening studies have estimated a β-thalassaemia trait carrier rate of up to 25%,^8,9,10^ while neighbouring countries such as Liberia and others in sub-Saharan Africa have reported estimation rates ranging from 9% to 20%.^11,12^ These estimates are commonly based on elevated HbA_2_ (> 3.5%) and HbF (> 1%) levels determined by high-performance liquid chromatography (HPLC). In contrast, earlier methods such as starch-block electrophoresis, used by Esan, reported a prevalence of < 1%, using a diagnostic cutoff of HbA_2_ > 3.8%.^13^
Sickle cell disease (SCD), which also results from a point mutation in the β-globin gene, may co-exist with β-thalassaemia. Among Nigerian patients with SCD, HPLC-based screening suggest a prevalence of co-inherited β-thalassaemia trait between 6% and 10%.^14,15^ However, a molecular-based study involving children under five in Northern Nigeria reported a much lower prevalence of 0.4% among children with SCD, and none among those without.^16^ The β-thalassaemia trait (β^+^-thal), when co-inherited with SCD, tends to ameliorate the clinical severity of SCD. Individuals with this compound heterozygous genotype (HbSβ^+^-thal) typically experience a milder disease course and fewer vaso-occlusive crises, similar to the HbSC genotype.^17^ Hence, it is expected to be at a higher prevalence in individuals with SCD compared to the general population, contrary to recent reports.
Diagnosis of β-thalassaemia trait in Nigeria commonly relies on HbA_2_ and HbF thresholds as the first initial screening process, but several confounding factors can elevate HbA_2_ levels, including α-thalassaemia,^16,18^ vitamin B12 or folate deficiency, antiretroviral therapy, and hyperthyroidism.^19^ Notably, α-thalassaemia itself is prevalent in Nigeria, with rates exceeding 35%.^18,20^ Nigeria bears the highest burden of SCD globally, yet the true prevalence and genetic characteristics of β-thalassaemia trait in this population remain uncertain. This study was therefore conducted to investigate the β-globin gene architecture in individuals with and without SCD in Nigeria by integrating molecular sequencing, variant annotation, and haplotype analysis to determine the prevalence and genetic spectrum of β-thalassaemia trait and to assess whether this differs significantly between the two groups. This study is among the first to use molecular genotyping to re-evaluate the prevalence of β-thalassaemia trait in Nigerians.
Methods
Ethical considerations
This study was approved by the University of Ibadan, University College Hospital, Ibadan Institutional Ethics Review Committee, College of Medicine, University of Ibadan (reference number: UI/EC/16/0195). Written consent form was obtained from each participant. The design and implementation of the research protocol were in accordance with the principles of The Declaration of Helsinki. A code was assigned to each participant and access to their demographic information stored on the computer was restricted to a few authorised individuals. This manuscript adheres to the Strengthening the Reporting of Observational Studies in Epidemiology guidelines for the reporting of observational studies.
Study design
This is a cross sectional study of 130 SCD patients in steady state attending University College Hospital Paediatric Outpatient Clinic and Haematology Clinic in Ibadan, Nigeria. This work is part of a pilot study on the genotypic characterisation of SCD patients in Ibadan, Nigeria carried out between September 2016 and March, 2018. The same number of apparently healthy individuals without SCD from the community were also recruited. Participants were aged 3 years – 69 years. Venous blood was collected from each participant and 3 mL was added to ethylenediaminetetraacetic acid (EDTA) bottles for complete blood count, HPLC, and DNA extraction at the Genetics and Bioethics Research Unit laboratory, College of Medicine, University of Ibadan, Nigeria. DNA amplification, sequencing and variant analysis were carried out at the Haematology-Oncology Laboratory, Department of Medicine, University of Chicago. More details on the demographic and clinical parameters, sampling, laboratory analyses, and DNA extraction from whole blood have been previously described.^18^ Samples from a subset of 26 individuals (from another health facility, Ring-Road State Hospital, Ibadan) with levels of HbA_2_ > 3% and/or HbF > 10% were genotyped along with the recruited participants because the haematological profiles were suggestive of β-thalassaemia trait.
Sample size determination
A total of 130 participants were recruited in each group (n = 260). The required sample size was estimated using the formula for comparing two proportions, assuming the prevalence of β-thalassaemia trait to be 6% among individuals with SCD and 20% among those without SCD. With a two-sided significance level (α) = 0.05 and 90% statistical power, the corresponding effect size (absolute difference) was 0.14. This calculation yielded a minimum of 116 participants per group, which was increased to 130 per group to accommodate potential non-response or data loss. At these parameters, the 95% confidence level and an estimated margin of error of ± 0.08 around the expected difference ensured adequate precision for detecting a statistically significant difference in β-thalassaemia trait prevalence between groups.
Exclusion criteria
Individuals with SCD who had undergone blood transfusion within three months of the study recruitment were excluded from enrolment.
Polymerase chain reaction amplification of the β-globin gene
The human β-globin gene is located on chromosome 11p15.4, GRCh38:5225464–5227071. It is 1608 base pairs (bp) long and contains three exons and two introns.^21^ This region was amplified in three separate polymerase chain reaction (PCR) reactions using three different sets of primers (Supplementary Table 1). The regions amplified by each set of primers are shown in Supplementary Figure 1. Each 15 µL PCR reaction contained 200 µM of each dNTP, 1.5 µL of 10x PCR buffer, 2.5 mM MgCl_2,_ 0.5 U Taq polymerase, 1.5 µL of 20 ng/µL of DNA template, and 10 µM of forward and reverse primers. PCR reactions were conducted in a thermocycler (PTC-200 Peltier Thermal Cycler; MJ Research Inc., Waltham, Massachusetts, United States) with an initial denaturation step at 98 °C for 2 min, followed by 35 cycles of 98 °C denaturation for 45 s, 63 °C annealing temperature for 45 s, and 72 °C extension for 1 min 30 s. A final 10-min extension at 72 °C was used to complete the reaction. A 5 µL aliquot of each amplicon was analysed by electrophoresis on a 1.5% agarose gel in tris-acetate EDTA buffer at 120 volts for 45 min. DNA samples from the 26 SCD patients from another study cohort were sequenced along with samples in this study. Only the first and third set of primers were used on this set of samples.
Diagnostic algorithm used to identify β-thalassaemia trait and analyse β-globin gene variants in the study population.
Cleaning of β-globin gene amplicons for sequencing
Amplicons were cleaned prior to sequencing. Each 10 µL reaction contained 1 µL Shrimp Alkaline Phosphatase Buffer (10x), 0.5 µL of 1 U/µL Shrimp Alkaline Phosphatase, 0.1 µL of 20 U/µL Exo 1, 3.4 µL of PCR grade water and 5 µL of amplicons. This was then incubated at 37 °C for 45 mins, 95 °C for 15 mins and 4 °C indefinitely. A 4 µL aliquot of the cleaned amplicons was submitted for Sanger sequencing.
Analysis of amplicon sequences
The sequences of the 1.6 kb β-globin gene amplicons were read and single nucleotide polymorphisms (SNPs) were detected using the Sequencher software version 4.2 (Gene Codes Corporation, Ann Arbor, Michigan, United States). The reference sequence of the GRCh38·p14 assembly with accession number NG_059281.1 was used as a template for sequence comparisons. The HbVar database^2^ was used to determine the rs numbers of the SNPs found within the amplicons, and dbSNP,^22^ ClinVar,^23^ Ensembl Variation database^24^ were used to determine the significance of each of the SNPs and allele frequencies in Africa and across the globe.
Haplotype analysis
Using Haploview version 4.2 (Broad Institute, Cambridge, Massachusetts, United States),^25^ the SNPs detected in individuals without SCD were analysed in one haplotype block since the full length of the amplicon is less than 2 kb. Haploview was used to determine haplotype frequencies and SNPs in linkage disequilibrium within the β-globin gene. The diagnostic testing strategy employed in this study is summarised in Figure 1.
Statistical analysis
Allele frequencies of SNPs detected in this study were compared with those found in the Ensembl 1000 Genomes Phase 3 project using Student’s t-test.
Results
Polymerase chain reaction amplicons of the β-globin gene
Three different sets of amplicons of size 774 bp, 645 bp and 574 bp were obtained from the PCR reactions (Supplementary Figure 2).
Haplotypes found within the β-globin gene, their frequencies and relationships generated by Haploview 4.2.
Single nucleotide polymorphisms in the β-globin gene
Eleven variants (SNPs and mutations) were found in the study population (Table 1). Ten SNPs were found in the non-SCD group. These include rs713040, rs33930165, rs334, rs111851677, rs10768683, rs7480526, rs7946748, rs1609812, rs537944366, and rs113152027. Some of the SNPs in the β-globin gene are in linkage disequilibrium. Worthy of note are the relationships between rs1609812, rs7946748, rs10768683 and rs713040, with relative linkage disequilibrium (D’) of 0.824 – 1.0 and r^2^ of 0.508 – 0.887 for each pair of the SNPs (Table 2), three of which were the only SNPs (rs713040, rs10768683 and rs1609812) found in each SCD patient. The 11th variant – rs33915217 was found in one patient in the separate SCD cohort.
Prevalence of β-thalassaemia in individuals with and without sickle cell disease
For this genetic analysis, participants with incomplete or inconclusive haematology or sequencing results were excluded. Results from 123 SCD patients, 117 non-SCD participants and the separate cohort of 26 SCD patients (n = 266) were analysed. No β-thalassaemia mutation was found in the SCD patients in the study main group, while a mutation that has been associated with benign β-thalassaemia trait (rs111851677) was found in three individuals of the non-SCD group in the heterozygous state, at a frequency of 0.014. These three individuals had normal haematocrit > 36%, HbF levels < 1%, and HbA_2_ levels < 3%. One of the individuals had a low mean corpuscular volume of 76.8% and an mean corpuscular haemoglobin of 23.8. One of the SCD patients in the separate cohort was found to have a moderately severe β-thalassaemia intermedia mutation (rs33915217C > A) (Supplementary Figure 3). This SNP is an intronic variant at chr11:5,226,925 (hg38). The patient had HbA at 8%, HbA_2_ at 4.8%, HbF at 11.9% and mean corpuscular volume at 67.9%. The full haematological parameters are also shown in Supplementary Figure 3.
Single nucleotide polymorphism haplotypes and their relationship
Eight haplotypes were discovered in the entire study population, including both SCD and non-SCD individuals. The haplotypes were grouped into two distinct ancestral haplotypes (Figure 2). The major ancestral haplotypes occurred at a total frequency of about 0.850 and the minor ancestral haplotypes occurred at a total frequency of 0.116 (Table 3). Four SNPs (rs713040, rs10768683, rs7946748 and rs1609812) differentiate the major and minor ancestral haplotypes, and they are in linkage disequilibrium (Table 2). Only one haplotype (TAGGAGAACG), a variant of the major ancestral haplotype, was found in all the participants with SCD in the homozygous state (HbSS) and another variant (TAGGAGATTG) was associated with haemoglobin C (HbC) (found in those with HbSC and HbAC genotype). The minor ancestral haplotype and its variants were found in 27 (24%) non-SCD participants in the heterozygous state; no participant had the homozygous state.
Discussion
The β-globin gene is highly polymorphic, harbouring numerous mutations that give rise to various haemoglobinopathies. These disorders are often population-specific, with unique combinations of mutations occurring in different regions of the world.^7,26^ β-thalassaemia, for instance, is caused by over 300 known mutations in the β-globin gene.^2,27^ In our genotyping study of 123 SCD patients in the main cohort, no known β-thalassaemia mutations were detected. Only one individual, an SCD patient in the separate sub-cohort, was found to carry a pathogenic β^+^ thalassaemia mutation (rs33915217C > A), which is typically associated with β-thalassaemia intermedia in the homozygous or compound heterozygous state. This sub-cohort was analysed because of elevated HbA_2_ and/or HbF levels, which in clinical practice often lead to presumptive diagnosis of β-thalassaemia trait based on HPLC alone.
The rs33915217 SNP has three rare alleles: C > A, C > G, and C > T^2^. All three disrupt normal splicing at the 5′ splice site, impairing mRNA processing and resulting in reduced or absent β-globin production.^2,28^ The A allele, found in our study, is particularly rare, and has previously been reported only in two individuals from Europe—one heterozygous and one homozygous.^28^ To our knowledge, this is the first report of this allele in both an African individual and an SCD patient. It was absent from the 1000 Genomes Phase 3 data set but is recorded at extremely low frequency (0.000009)^24^ in the gnomAD exome (non-Finnish European) data set.
The rs111851677 variant (IVS-I-108), found in three individuals without SCD in the heterozygous state, has been previously reported in β-thalassaemia carriers from Cuba,^29^ Iran,^30^ and Greece.^31^ Though its clinical impact is debated, it may impair RNA splicing because of its location near the 3′ splice site.^29,32^ In our study, however, individuals carrying this variant showed normal haematological profiles, suggesting a benign role.
Our findings support earlier studies from Nigeria^13,16^ that reported β-thalassaemia mutations to be rare (< 1%) in the population. This contrasts with recent reports suggesting a prevalence of 6% to 10% in SCD patients and 20% to 25% in the general Nigerian population.^9,10,14,15^ The discrepancy likely stems from methodological differences, as these studies relied solely on haematologic indices and HPLC thresholds (HbA_2_ > 3.0% – 3.5%, HbF > 1%) to infer β-thalassaemia trait without molecular confirmation. Such screening-based approaches are prone to overestimation because of conditions that elevate HbA_2_, including α-thalassaemia,^16,18^ vitamin B_12_ or folate deficiency, antiretroviral therapy, and thyroid disorders.^19^ In our study, all the individuals (except one) in both the main and separate cohorts with elevated HbA_2_ levels had no detectable β-thalassaemia mutations on genetic testing. Similar findings were reported by Inusa et al.,^16^ who found no underlying β-thalassaemia mutations despite elevated HbA_2_ levels (up to 7.4%). This underscores the need for standardised diagnostic methods integrating molecular genotyping.
Given the high prevalence of α thalassaemia in Nigeria (estimated at 35% – 45%),^18,20^ this could contribute significantly to spurious elevations in HbA_2_ and possible misdiagnosis of β-thalassaemia trait. Interestingly, since β-thalassaemia trait can ameliorate SCD symptoms, its frequency would be expected to be higher in SCD patients than in the general population. However, recent HPLC-based studies report the reverse,^9,10^ further emphasising the limitations of relying solely on haematologic indices for diagnosis.
Apart from rs33915217 (β-thalassaemia), rs334 (HbS), and rs33930165 (HbC), all other variants identified in our study were non-pathogenic intronic SNPs. Notably, rs713040G—which has a higher global frequency than its reference allele—was found in 100% of SCD patients, compared to ~80% in non-SCD, African, and global populations. Similarly, rs33930165 (HbC mutation) was observed in 1.8% of the non-SCD cohort, aligning with its expected prevalence in African populations (0.013–0.028).^24^ The rs334 (HbS mutation) had a frequency of 0.085 in the non-SCD group, which is consistent with African averages and lower than in the Yoruba in Ibadan reference population.
Other intronic variants found in this study (rs10768683, rs7480526, rs7946748, rs113152027, rs1609812) displayed population frequencies consistent with African ancestry. One rare 3′ untranslated region variant, rs537944366, was detected in only one individual. This variant, absent in the Yoruba in Ibadan and only recorded in Mende in Sierra Leone and gnomAD non-Finnish Europeans, is being reported for the first time in a Nigerian (Yoruba) population.
Our haplotype analysis revealed two ancestral haplotypes: a major haplotype (frequency: 0.850), typical of African populations, and a minor haplotype (frequency: 0.116), atypical for Africans and more common in South and East Asian populations. These haplotypes are differentiated by four SNPs in linkage disequilibrium (rs713040, rs10768683, rs7946748, rs1609812). The minor ancestral haplotype and its variants were found in 27 (24%) non-SCD participants in the heterozygous state; no participant had the homozygous state. Hardy-Weinberg expectations suggest a frequency too low to detect given the current sample size.
Only the major haplotype (TAGGAGAACG) was associated with HbS and HbC mutations. Interestingly, the SCD patient with the β-thalassaemia intermedia mutation was heterozygous for both haplotypes, linking the pathogenic variant to the minor ancestral haplotype. The rs33915217 β-thalassaemia mutation was found on the minor haplotype, supporting the idea of genetic admixture or historical gene flow into this population. This pattern suggests that HbS and HbC mutations in this population have likely evolved on a common African haplotypic background and have undergone minimal recombination.
Our findings challenge the assumption of a high prevalence of β-thalassaemia trait in Nigeria. The findings also validate the robustness of the sequencing and analysis pipeline. Seven SNPs from our data showed the same linkage disequilibrium pattern in Yoruba in Ibadan individuals from the 1000 Genomes Project,^33^ reinforcing the accuracy of our haplotype deductions.
Limitations
The study population is from a specific region of the country that is predominantly of Yoruba ethnicity, which may not represent the full genetic diversity of the Nigerian population. The sample size, although statistically adequate for detecting group differences, may not have been large enough to detect very rare β-thalassaemia variants. Only the β-globin gene region was sequenced; hence, mutations in regulatory or flanking regions may have been missed.
Conclusion
This study demonstrates that β-thalassaemia trait could be rare in both SCD and non-SCD individuals in the Yoruba population of Nigeria, and is associated with a minor ancestral haplotype likely introduced through genetic admixture. Haematologic parameters such as elevated HbA_2_ and HbF, while being useful screening tools, are insufficient for the definitive diagnosis of β-thalassaemia trait in African populations because of confounding factors such as α-thalassaemia, infections, and folate deficiency. Genetic confirmation is essential to avoid misdiagnosis, especially in regions with high prevalence of SCD, where correct genotyping is crucial for accurate disease classification, appropriate clinical management, and genetic counseling.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Cao A, Galanello R, Rosatelli MC. Genotype-phenotype correlations in beta-thalassaemias. Blood Rev. 1994;8(1):1–12. 10.1016/0268-960X(94)90002-78205005 · doi ↗ · pubmed ↗
- 2A database of human haemoglobin variants and thalassaemias [homepage on the Internet]. 2024 [cited 2024 Jun 17]. Available from: https://globin.bx.psu.edu/cgi-bin/hbvar/query_vars 3
- 3Cao A, Moi P. Regulation of the globin genes. Pediatr Res. 2002;51(4):415–421. 10.1203/00006450-200204000-0000311919323 · doi ↗ · pubmed ↗
- 4Weatherall DJ. Phenotype-genotype relationships in monogenic disease: Lessons from the thalassaemias. Nat Rev Genet. 2001;2(4):245–255. 10.1038/3506604811283697 · doi ↗ · pubmed ↗
- 5Galanello R, Origa R. Beta-thalassaemia. Orphanet J Rare Dis. 2010;5:11. 10.1186/1750-1172-5-1120492708 PMC 2893117 · doi ↗ · pubmed ↗
- 6Kattamis A, Forni GL, Aydinok Y, Viprakasit V. Changing patterns in the epidemiology of β-thalassaemia. Eur J Haematol. 2020;105(6):692–703. 10.1111/ejh.1351232886826 PMC 7692954 · doi ↗ · pubmed ↗
- 7Flint J, Harding RM, Boyce AJ, Clegg JB. The population genetics of the haemoglobinopathies. Baillieres Clin Haematol. 1998;11(1):1–51. 10.1016/S 0950-3536(98)80069-310872472 · doi ↗ · pubmed ↗
- 8Kotila T. Beta thalassaemia in Nigeria: Myth or fact? Afr J Med Med Sci. 2013;42(3):261–264.24579388 · pubmed ↗