High-quality genome assembly and annotation of the white-cheeked goby, Rhinogobius duospilus (Herre, 1935) (Gobiiformes: Oxudercidae)
Lixian Wu, Jiantao Hu, Shanshuo Zhang, Yi Qing Fam, Jianhong Xia, Chenhong Li

TL;DR
This paper presents a high-quality genome assembly for the white-cheeked goby, a small fish native to southern China and Vietnam, aiding future studies on its evolution and adaptation.
Contribution
The first chromosome-level genome assembly of Rhinogobius duospilus using PacBio HiFi and Hi-C technologies.
Findings
A chromosome-level genome assembly of 1,031.61 Mb with 96.14% completeness was achieved.
24,418 protein-coding genes were predicted, with 96.8% successfully annotated.
The genome provides a resource for studying population differentiation and adaptive evolution in R. duospilus.
Abstract
The white-cheeked goby (Rhinogobius duospilus) is a small stream-dwelling fish endemic to southern China and Vietnam. With a sucker-like modified pelvic fin that helps it cling to substrate in fast-flowing water and vibrant breeding colors in males, R. duospilus is particularly appealing to aquarium enthusiasts. To investigate its distribution patterns, evolutionary history, and molecular adaptations to local environments, a high-quality genome assembly is critically needed. By employing PacBio HiFi sequencing combined with Hi-C-assisted assembly technology, we successfully obtained a chromosome-level genome assembly of R. duospilus. The final assembly yielded a genome size of 1,031.61 Mb with a scaffold N50 of 45.55 Mb. Approximately 991.84 Mb of genomic sequence was anchored onto 22 chromosome pairs. Benchmarking Universal Single-Copy Orthologs assessment indicated high genome…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Fig. 1
Fig. 1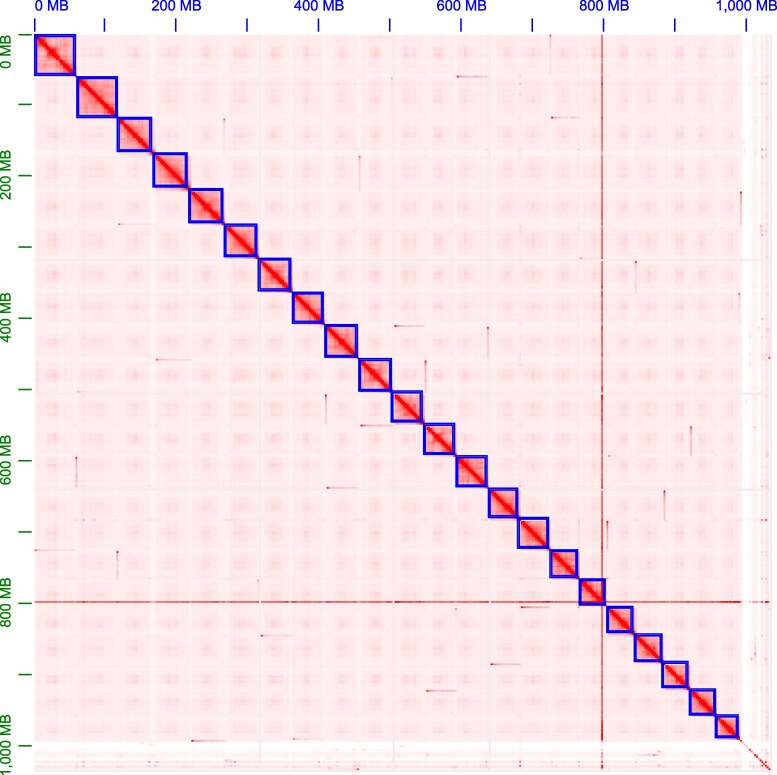 Fig. 2
Fig. 2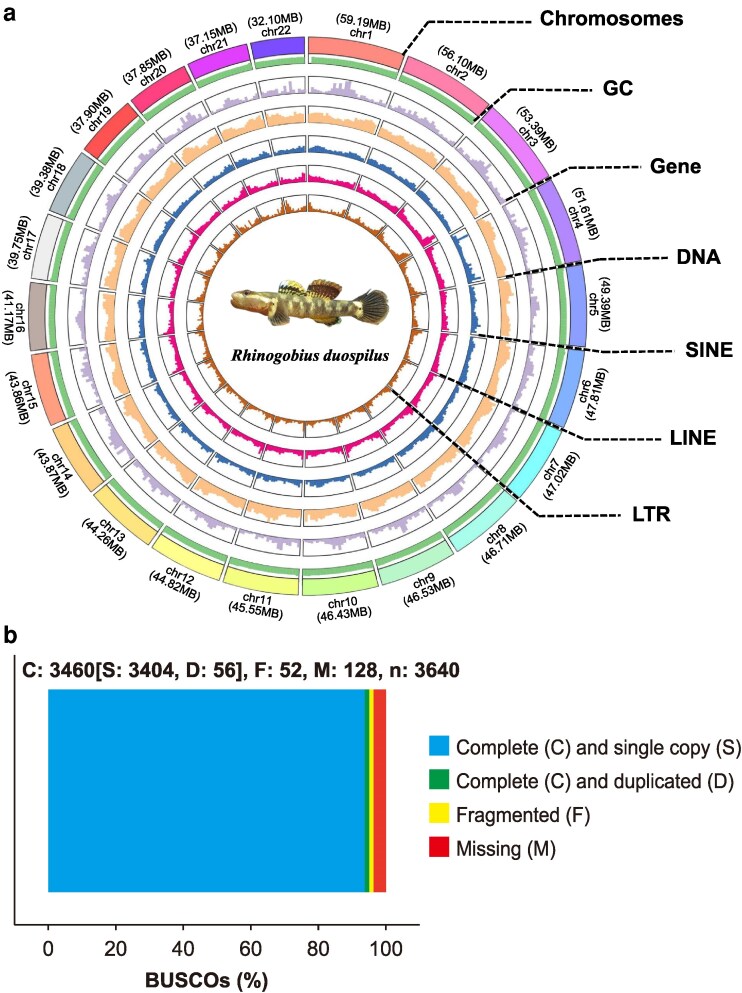 Fig. 3
Fig. 3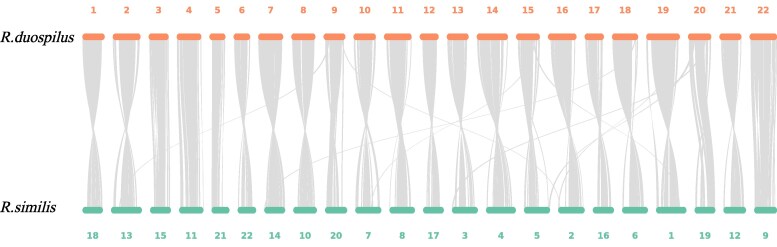 Fig. 4
Fig. 4| Item | Description |
|---|---|
| Investigation type | Vertebrate complete genome sequence |
| Project name | PRJNA1273456 |
| Geographic location (latitude and longitude) | 22.66 N, 113.97 E |
| Geographic location (country and/or sea, region) | China: Guangdong, Shenzhen |
| Collection date | 2024 September 1 |
| Broad-scale environmental context | Aquatic biome ENVO_00002030 |
| Local environmental context | Freshwater river biome ENVO_01000253 |
| Environmental medium | Freshwater ENVO_00002011 |
| Sequencing method | PacBio HiFi sequencing Hi-C library |
| Assembly | hifiasm v0.16.0 Trinity v2.11.0 |
| Attribute | Haplotype 1 | Haplotype 2 |
|---|---|---|
| Genome size | 1.44 Gb | 1.35 Gb |
| Scaffold N50 | 6.41 Mb | 6.80 Mb |
| Number of scaffolds | 2,491 | 1,311 |
| Longest scaffold | 33.81 Mb | 40.82 Mb |
| GC content | 40.60% | 40.49% |
| Attribute | Value |
|---|---|
| Size | 1,031.61 Mb |
| Chromosome | 22 |
| Chromosome % | 96.14% |
| Complete BUSCO (Actinopterygii odb10) | 95% |
| Protein-coding genes | 24,418 |
| Annotated % | 96.80% |
- —National Key Research and Development Program of China10.13039/501100012166
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenomics and Phylogenetic Studies · Developmental Biology and Gene Regulation · Ichthyology and Marine Biology
Introduction
The white-cheeked goby (Rhinogobius duospilus) is a small benthic fish endemic to subtropical creeks in China and Vietnam (Pan et al. 1991). Ecologically, this species plays an essential role in the energy cycle of the freshwater ecosystem, feeding on small invertebrates while serving as prey for larger carnivorous species (Pan et al. 1991). Recently, R. duospilus has gained popularity among aquarium enthusiasts due to its specialized sucker-like pelvic fin, which allows it to cling to substrate in flowing water, as well as the vibrant breeding colors exhibited by mature males. Unlike other amphidromous euryhaline congeners, R. duospilus is a freshwater-dwelling species (Dudgeon and Corlett 2004; Ho and Dudgeon 2016). Such ecological habit results in highly fragmented habitats of this species, leading to genetic differentiation among populations across distinct environments (Wu et al. 2016). The most conspicuous manifestation of such differentiation lies in the morphological diversity observed between populations, particularly in body color polymorphism. This phenotypic variation poses challenges for taxonomy and precise management of population genetic resources of R. duospilus.
Currently, genetic studies on R. duospilus have been limited to local populations using restricted molecular markers (Wu et al. 2016). Although these studies have revealed interesting local population structure, significant knowledge gaps remain concerning the species' genome-wide genetic diversity and range-wide population structure. Moreover, the genetic basis underlying the observed morphological polymorphisms in R. duospilus has yet to be elucidated. Advances in high-throughput sequencing and assembly technologies have enabled genome studies to expand beyond evolution into areas such as gene function, ecology, and environmental adaptation. In light of these gaps, genome sequence with high-quality assembly and annotation of R. duospilus is critically needed. A well-characterized genome will serve as a foundational resource for elucidating the genetic mechanisms underlying population differentiation, morphological variation, and adaptive evolution through comparative genomics. Such data will significantly advance taxonomic classification, evolutionary research, and conservation genetics for this species.
Materials and methods
Sample collection, library preparation, and sequencing
A male R. duospilus (Fig. 1) with the standard length of 50 mm was collected from near its type locality in Shenzhen City, Guangdong Province, China, on 2024 September 1 (Table 1). From this specimen, we collected liver tissue for PacBio long-read sequencing and muscle tissue for Hi-C library construction and sequencing. In this process, the SMRTbell long-read sequencing library was constructed following the official protocol of the SMRTbell Template Prep Kit and sequenced on the PacBio Sequel II platform. Meanwhile, the Hi-C library was prepared using the restriction enzyme DpnII for digestion combined with formaldehyde cross-linking. The completed Hi-C library was sequenced on an Illumina sequencing platform by Novogene (Beijing, China). Furthermore, 8 additional tissue types were dissected for transcriptome analysis: eye, brain, gill, muscle, skin, liver, heart, and spleen. The RNA-seq libraries constructed from these tissue samples were sequenced on the Illumina platform as well. All library construction and sequencing procedures followed the manufacturer’s specified protocols using official reagent kits to ensure data quality and reproducibility.
The specimen of R. duospilus.
Genome assembly of R. duospilus
Using the hifiasm v0.16.0 software (https://github.com/chhylp123/hifiasm) (Nishii et al. 2023), a preliminary genome assembly was performed with PacBio HiFi sequencing data. The assembled results were then indexed using BWA v0.7.17 (https://github.com/sghignone/bwa) (Li 2013) for subsequent analysis. Additionally, DpnII restriction enzyme cutting site information was generated for the genome, followed by executing an awk command to obtain length information for each sequence.
Subsequently, the Juicer v1.6 software (https://github.com/aidenlab/juicer.git) (Durand et al. 2016b) was employed to process Hi-C data, utilizing the preliminary genome assembly from hifiasm, the restriction enzyme cutting site information, and the sequence length information. This step generated an interaction matrix file for further assembly.
Using 3D-DNA v180922 (https://github.com/aidenlab/3d-dna.git) (Dudchenko et al. 2017), the genome sequence file and the interaction matrix produced by Juicer were combined to scaffold the genome sequences onto chromosomes, achieving chromosome-level genome assembly (parameter: r = 5). Juicebox v1.11.08 (https://github.com/theaidenlab/juicebox/wiki) (Durand et al. 2016a) was then used to manually correct scaffolding errors, thereby improving assembly accuracy. Finally, a Python script (https://github.com/luliangBio/3d-dna_tools) was called to integrate the results from Juicebox and hifiasm, generating the final chromosomal genome sequence.
The quality and completeness of the genome were assessed using Benchmarking Universal Single-Copy Orthologs (BUSCO) v5.5.0 (https://github.com/robsyme/busco.git) (Simão et al. 2015). A database named actinopterygii_odb10 was used for BUSCO analysis. The BUSCO database serves as a standardized, comprehensive, and reliable tool for effectively evaluating genome assembly completeness and annotation accuracy.
Transcriptome assembly
Using the assembled genome of R. duospilus as the reference genome, BWA v0.7.17 (Li 2013) was employed to construct an index for the genome. Subsequently, the same software was used to align the transcriptome data to the reference genome, generating files in sam format. These sam files were then converted into sorted binary files in .bam format using SAMTools v0.1.19 (https://www.htslib.org/workflow) (Li et al. 2009) to facilitate downstream analysis. For the transcriptome assembly step, the Trinity v2.11.0 (https://github.com/trinityrnaseq/trinityrnaseq.git) software (Haas et al. 2013) was used to perform reference-based transcriptome assembly based on the bam files for sequences from 8 different tissues. The assembled sequences from these 8 tissues were then merged, and cd-hit (Li and Godzik 2006) was applied to remove redundancy from the merged Trinity assembly results. This process yielded a comprehensive transcriptome assembly, which was subsequently used for genome annotation.
Gene prediction and annotation
Using RepeatMasker v4.1.0 (https://www.repeatmasker.org/), the genome was soft-masked by de novo repeat sequence database construction. Subsequently, protein databases of 6 closely related species were downloaded from the National Center for Biotechnology Information (NCBI), including Periophthalmus magnuspinnatus (GCA_009829125), Boleophthalmus pectinirostris (GCA_026225935), Mugilogobius chulae (GCA_046056395), Pomatoschistus minutus (GCA_009829125), Eucyclogobius newberryi (GCA_026437365), and Danio rerio (GCA_049306965). The protein sequences of these species were merged and deduplicated. The BRAKER3 pipeline (https://github.com/Gaius-Augustus/BRAKER.git) (Gabriel et al. 2024) was then employed to predict and annotate the genome of R. duospilus, integrating the soft-masked genome, the deduplicated protein database of related species, and the assembled transcriptome data. Finally, InterProScan (https://interproscan-docs.readthedocs.io/en/v5/) was used to analyze and summarize the annotation results. InterProScan integrates multiple databases. It is a powerful biological sequence analysis tool that consolidates various protein signature databases and predictive tools, such as GO, PANTHER, SMART, SUPERFAMILY, Gene3D, Pfam, ProSiteProfiles, and FunFam, among others.
Mitochondrial genome assembly and annotation
The mitochondrial genome of R. duospilus was assembled using NOVOPlasty 4.3.1 (https://github.com/ndierckx/NOVOPlasty) with the COI gene of Rhinogobius similis as the seed. The coding sequence (CDS) were then annotated in Geneious (https://www.geneious.com/) using the complete mitochondrial genome of R. similis as a reference. Finally, annotations for rRNA, tRNA, and the D-loop control region were added by incorporating predictions from MITOS2 (https://gitlab.com/Bernt/MITOS/), yielding the complete mitochondrial genome annotation.
Genome synteny analysis
The genome assembly and annotation files of R. similis were downloaded from the NCBI database (accession number: GCA_019453435.1). Chromosomal synteny analysis between R. duospilus and R. similis was then performed using MCScanX (https://github.com/wyp1125/MCScanX) under default parameters.
Results and discussion
Sequencing and genome assembly results
The PacBio HiFi sequencing achieved 100× coverage, which is sufficient for chromosome-level genome assembly. The HiFi sequencing data consisted of 51.5 Gb total base pairs from 3,145,229 reads, with maximum and mean read lengths of 59,675 bp and 16,374 bp, respectively, and a read length N50 of 16,291 bp. Additionally, the Hi-C data reached 100× coverage, indicating that the obtained Hi-C sequencing data were adequate for chromosome anchoring. The RNA-seq read length was 150 bp.
The statistical results of the 2 haplotypes obtained by hifiasm assembly are presented in Table 2. The final assembled genome size was 1,031.61 Mb, with approximately 991.84 Mb (96.14%) of the sequences anchored to 22 chromosomes. The Hi-C heatmap is shown in Fig. 2. The undetermined sequences (N) accounted for only 0.02%, indicating a high-quality chromosome-level assembly of R. duospilus with superior accuracy. Furthermore, the high contig N50 (6.58 Mb) and scaffold N50 (45.55 Mb) values demonstrate excellent assembly quality. Notably, the scaffold N50 was significantly higher than the contig N50, indicating effective joining of multiple contigs and suggesting high sequence continuity in the assembly. The detailed assembly statistics are presented in Table 3, with the lengths of individual assembled chromosomes shown in Fig. 3a.
Hi-C map of R. duospilus.
Chromosome-level assembly of R. duospilus. a) Circos plot of the R. duospilus genome, with visualization of chromosome sizes, GC content, gene density, DNA, SINE, LINE, and LTR in order from outside to inside. b) BUSCO evaluation results of the R. duospilus genome assembly.
The detailed BUSCO evaluation results of assembly are presented in Fig. 3b. The Complete BUSCOs accounted for 95% of the dataset, exceeding the 90% benchmark and slightly higher than the Complete BUSCO score (93.02%) reported for the congeneric species R. similis (Hu et al. 2022). These results demonstrate excellent genome completeness in R. duospilus assembly, confirming the high-quality genome assembly we achieved for R. duospilus.
Gene prediction and annotation
A total of 24,418 genes were predicted, of which 23,660 (96.8%) were successfully annotated, indicating that the vast majority of gene sequences were matched to known databases. The assembled R. duospilus genome achieved consistently high annotation rates in 5 of the reference databases examined, including GO, PANTHER, SUPERFAMILY, Gene3D, and Pfam. The PANTHER database yielded the highest success rate at 89.9%, followed by Pfam at 85.2%, GO at 80.2%, Gene3D at 73.5%, and SUPERFAMILY at 68.9%.
Mitochondrial genome assembly and annotation
The complete mitochondrial genome of R. duospilus was successfully assembled using NOVOPlasty 4.3.1. The genome is a circular DNA molecule with a total length of 16,485 bp. Annotation results revealed that it contains 13 protein-coding genes, 2 rRNAs, 22 tRNAs, and 1 D-loop control region. The gene composition and arrangement are consistent with the typical mitochondrial genome structure of vertebrates.
Genome synteny analysis
The synteny analysis between R. duospilus and R. similis is shown in Fig. 4, revealing a high degree of synteny between 2 species. Chromosome fusion and fission events were observed, such as chromosome 9 of R. duospilus corresponding to chromosomes 13, 20, and 2 of R. similis. Similarly, chromosome 15 of R. duospilus corresponds to chromosomes 7, 5, and 1 of R. similis; chromosome 18 of R. duospilus corresponds to chromosomes 14 and 6 of R. similis; and chromosome 20 of R. duospilus corresponds to chromosomes 3, 5, 2, and 19 of R. similis.
Synteny analysis between R. duospilus and R. similis.
Conclusion
This study utilized PacBio HiFi sequencing technology and Hi-C data-assisted assembly to complete a chromosome-level genome assembly of the R. duospilus. Through genome assembly and gene prediction annotation, the research revealed the genome size and functional characteristics of genes. This study not only provides crucial genomic data for classification and evolutionary research of R. duospilus but also offers new insights into understanding the mechanisms of population differentiation and adaptation in this species.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Dudchenko O et al 2017. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science. 356:92–95. 10.1126/science.aal 3327.28336562 PMC 5635820 · doi ↗ · pubmed ↗
- 2Dudgeon D, Corlett R. 2004. The ecology and biodiversity of Hong Kong. Joint Publishing (Hong Kong) Ltd.
- 3Durand NC et al 2016 a. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 3:99–101. 10.1016/j.cels.2015.07.012.27467250 PMC 5596920 · doi ↗ · pubmed ↗
- 4Durand NC et al 2016 b. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3:95–98. 10.1016/j.cels.2016.07.002.27467249 PMC 5846465 · doi ↗ · pubmed ↗
- 5Gabriel L et al 2024. BRAKER 3: Fully automated genome annotation using RNA-seq and protein evidence with Gene Mark-ETP, AUGUSTUS and TSEBRA. Genome Res. 34:769–777. 10.1101/gr.278090.123.38866550 PMC 11216308 · doi ↗ · pubmed ↗
- 6Haas BJ et al 2013. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc. 8:1494–1512. 10.1038/nprot.2013.084.23845962 PMC 3875132 · doi ↗ · pubmed ↗
- 7Ho BS, Dudgeon D. 2016. Movement of three stream-resident balitoroid loaches and a goby in a Hong Kong hillstream. Ecol Freshw Fish. 25:622–630. 10.1111/eff.12240. · doi ↗
- 8Hu Y et al 2022. A high-resolution genome of an euryhaline and eurythermal rhinogoby (Rhinogobius similis Gill 1895). G 3 (Bethesda). 12:jkab 395. 10.1093/g 3journal/jkab 395.34792546 PMC 9210307 · doi ↗ · pubmed ↗