Pyrimidine-containing natural products: occurrences and biological activities
Jian-Neng Yao, Yongjie Zhu, He-Ping Chen, Yihua Chen

TL;DR
This review explores pyrimidine-containing natural products and their diverse biological activities, aiming to highlight their potential for drug development.
Contribution
The paper provides a comprehensive survey of pyrimidine-containing natural products from 2004 to 2025, emphasizing their structural diversity and therapeutic potential.
Findings
156 pyrimidine-containing compounds were analyzed for their structural and biological features.
These compounds show a wide range of activities, including anticancer, antiviral, and anti-inflammatory effects.
The review highlights the potential of these natural products for lead compound optimization and drug development.
Abstract
Nitrogen-containing core structure pyrimidine has long been regarded as one of the privileged scaffolds for novel drug development. Natural products embedded with pyrimidine motifs are distinguished by their exceptional scaffold diversity and vast structural complexity, which endow them with versatile biological activities, including anticancer, antiviral, antifungal and anti-inflammatory activities. This review is dedicated to surveying a series of structurally distinctive naturally occurring compounds characterized by the presence of an aromatic heterocyclic pyrimidine moiety covering from 2004 to early 2025. Multiple key aspects of these 156 pyrimidine-containing compounds, including natural sources, features of chemical structure, biological activities, as well as biosynthetic studies are summarized. The review emphasizes the enduring potential of natural products, highlighting…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
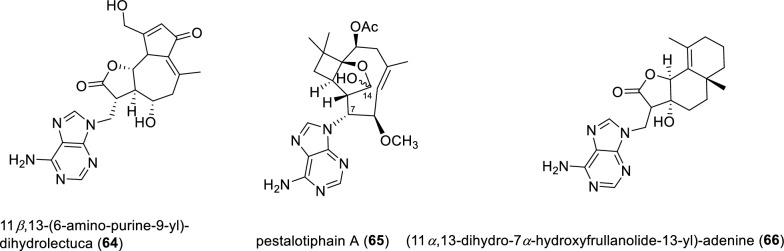 Figure 10
Figure 10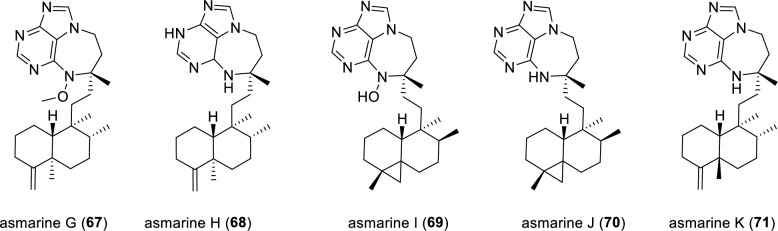 Figure 11
Figure 11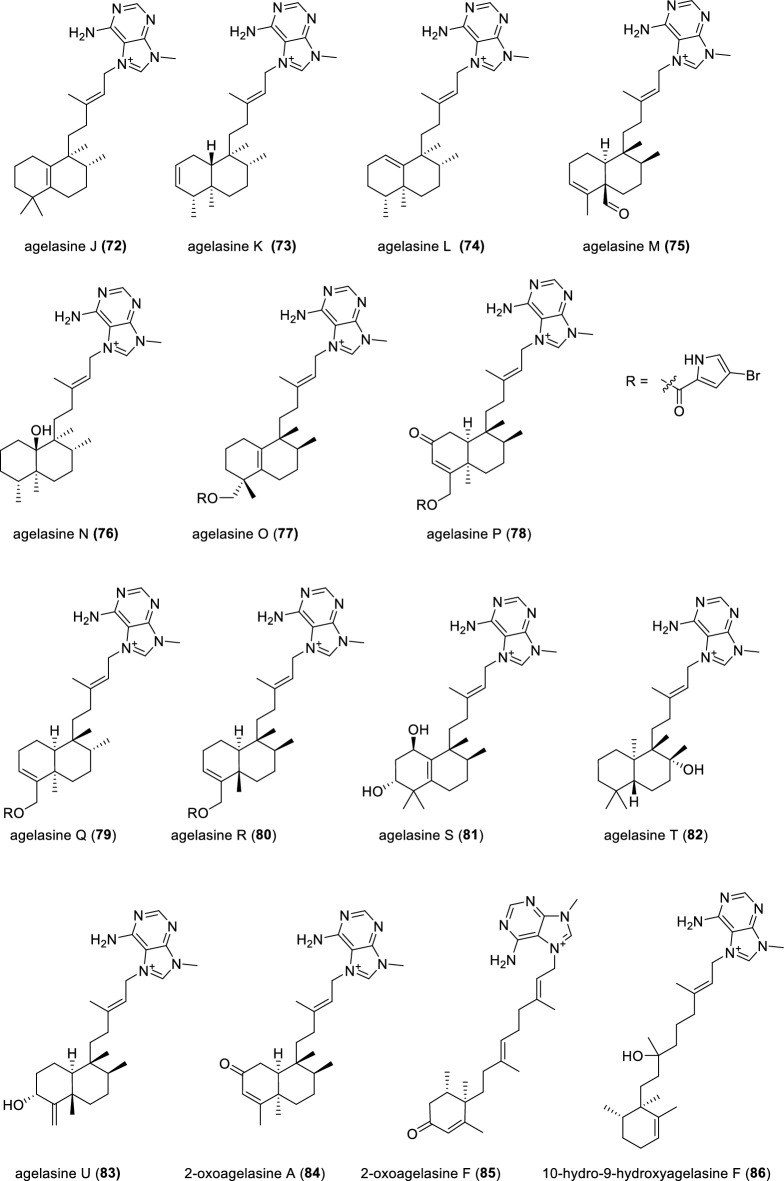 Figure 12
Figure 12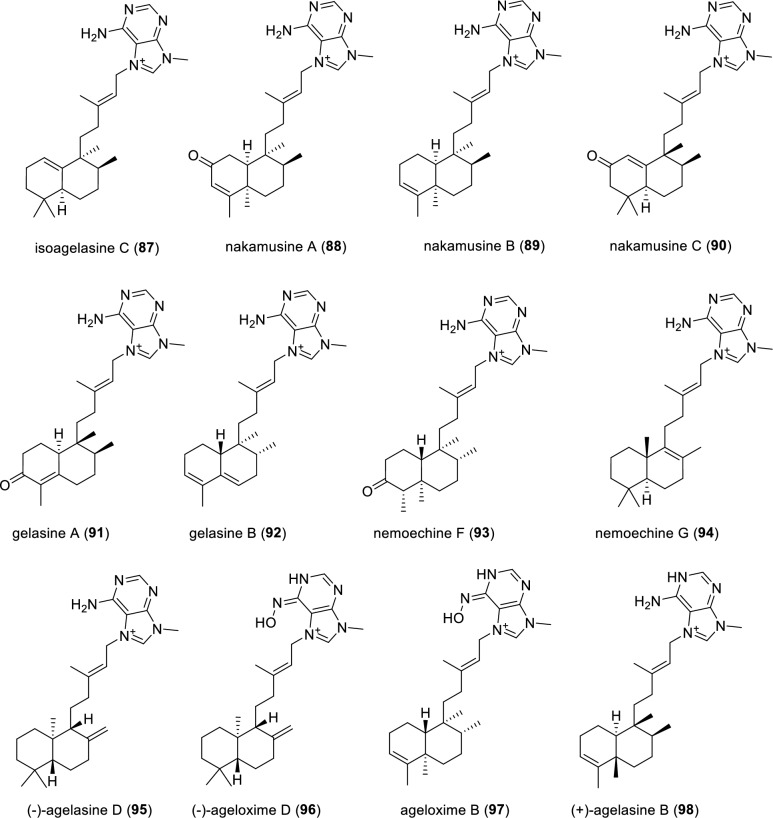 Figure 13
Figure 13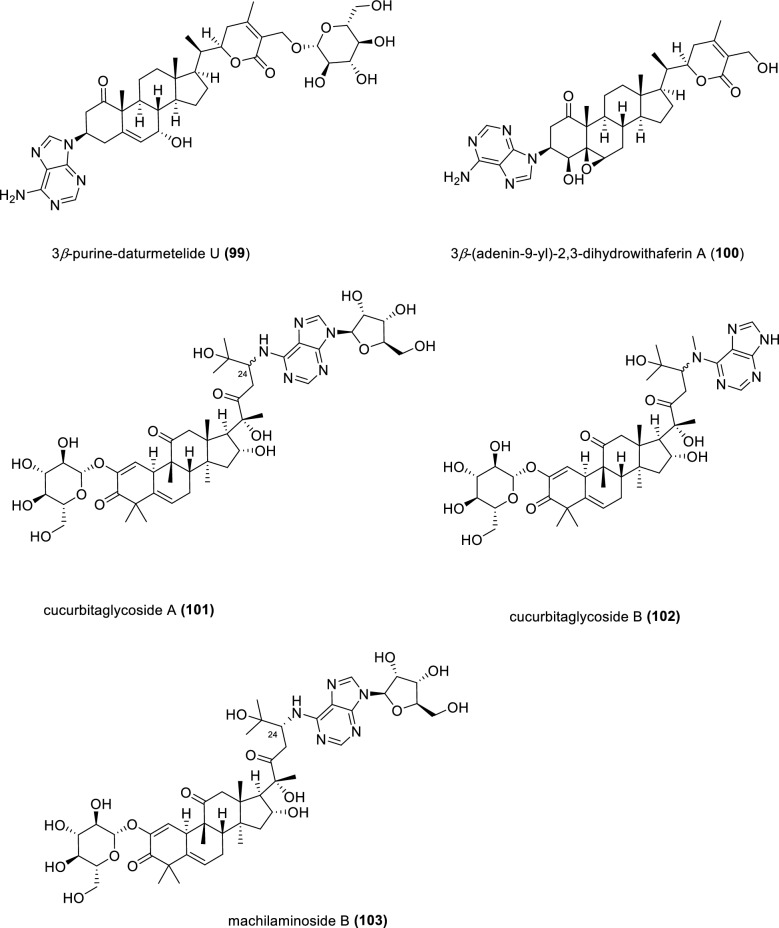 Figure 14
Figure 14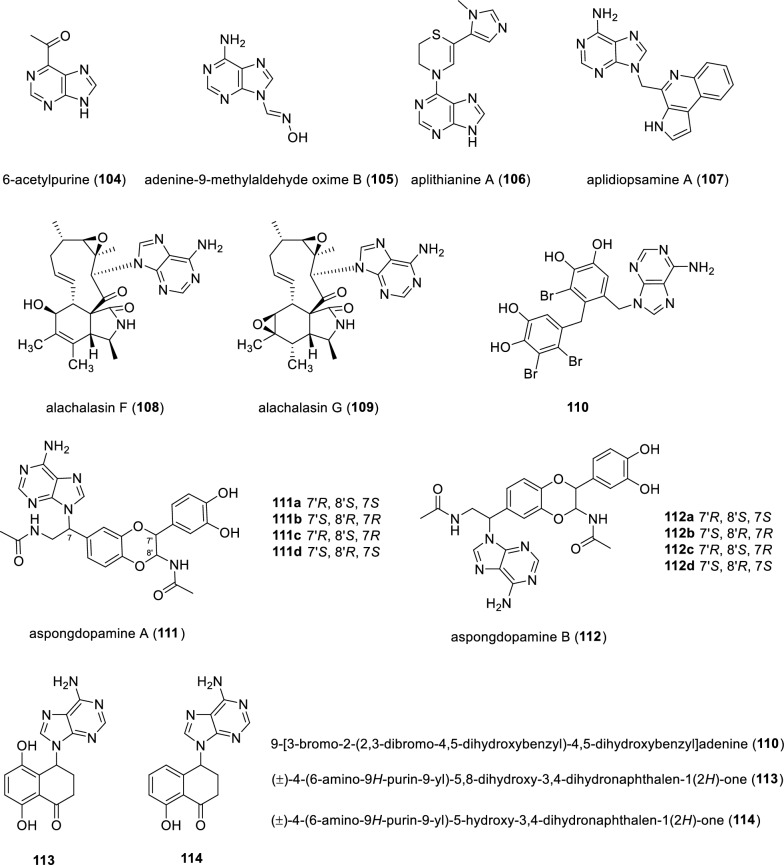 Figure 15
Figure 15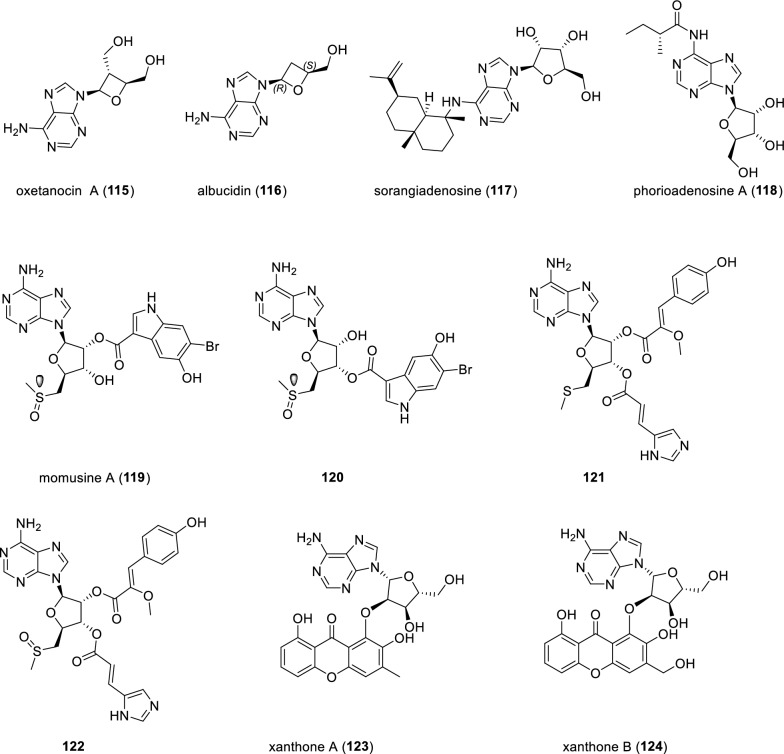 Figure 16
Figure 16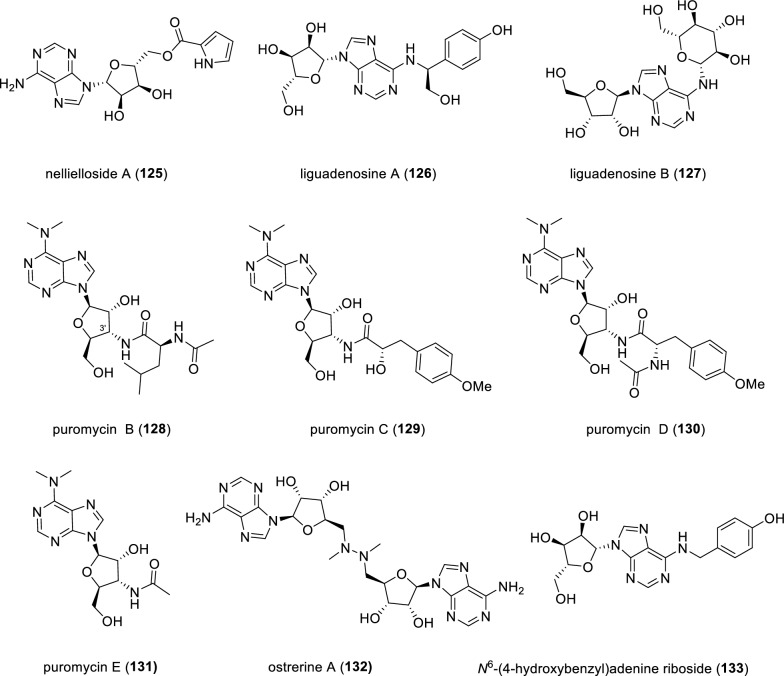 Figure 17
Figure 17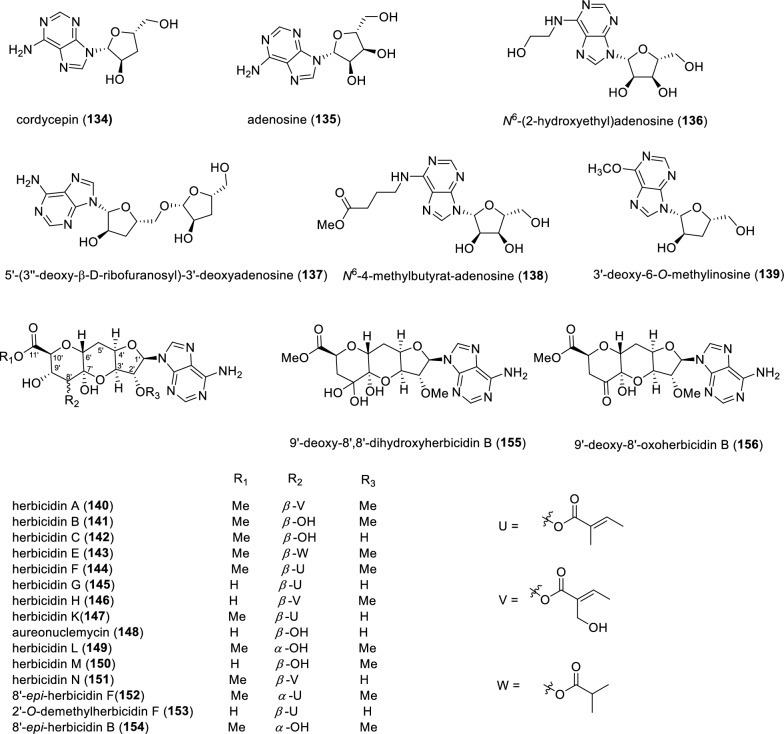 Figure 18
Figure 18 Figure 1
Figure 1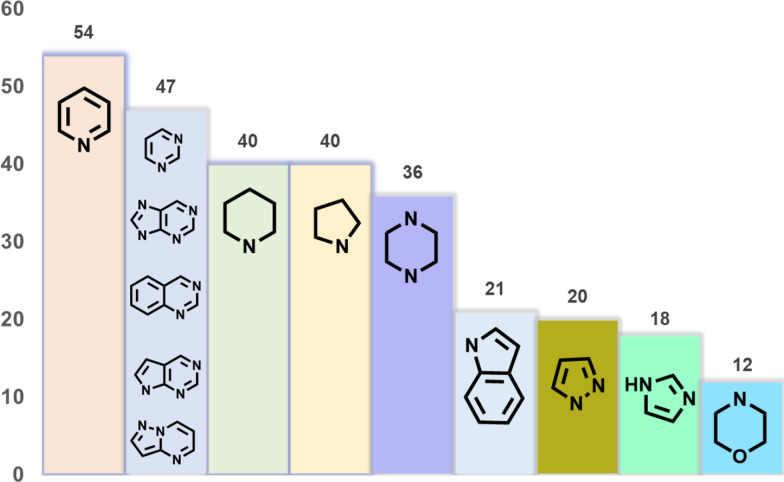 Figure 2
Figure 2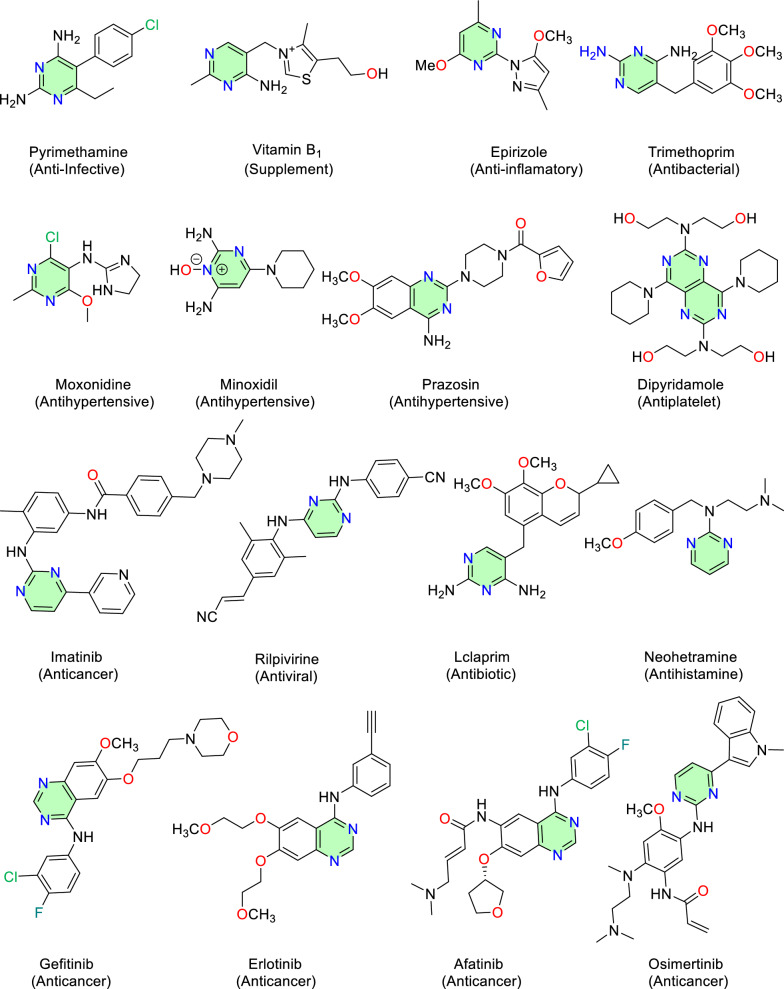 Figure 3
Figure 3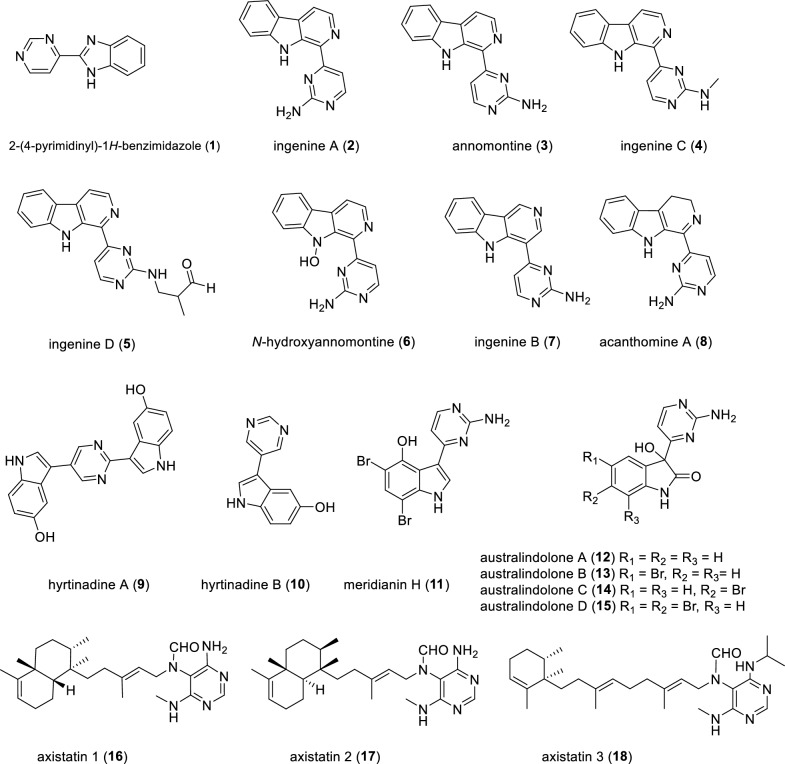 Figure 4
Figure 4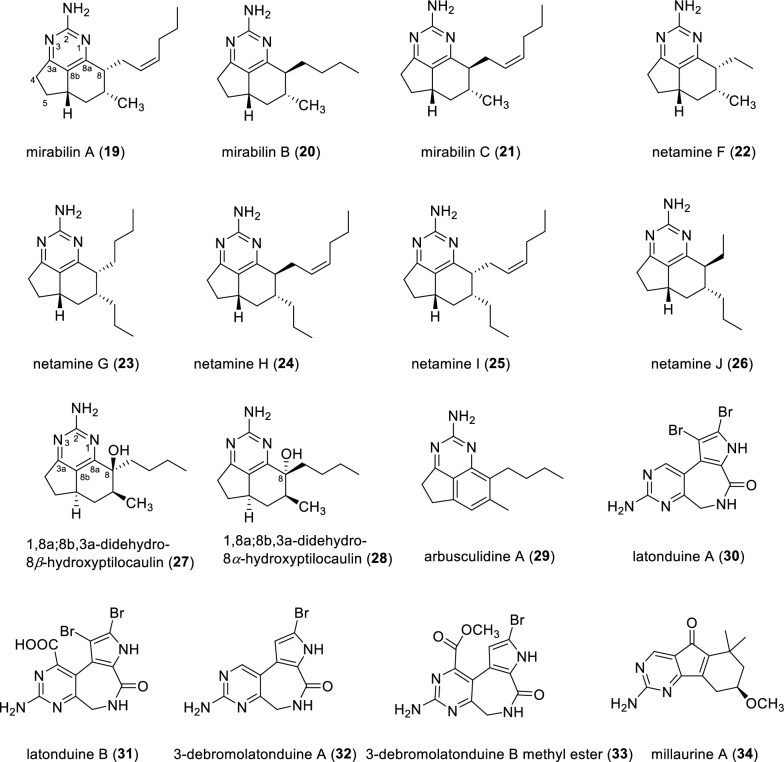 Figure 5
Figure 5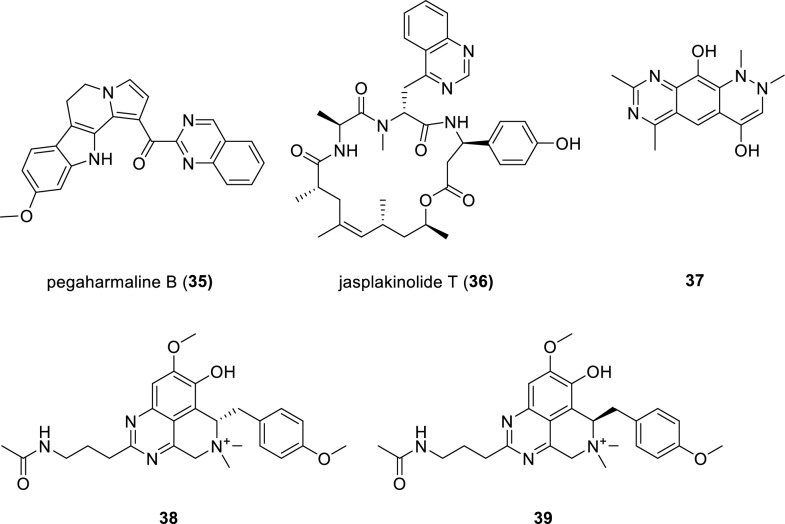 Figure 6
Figure 6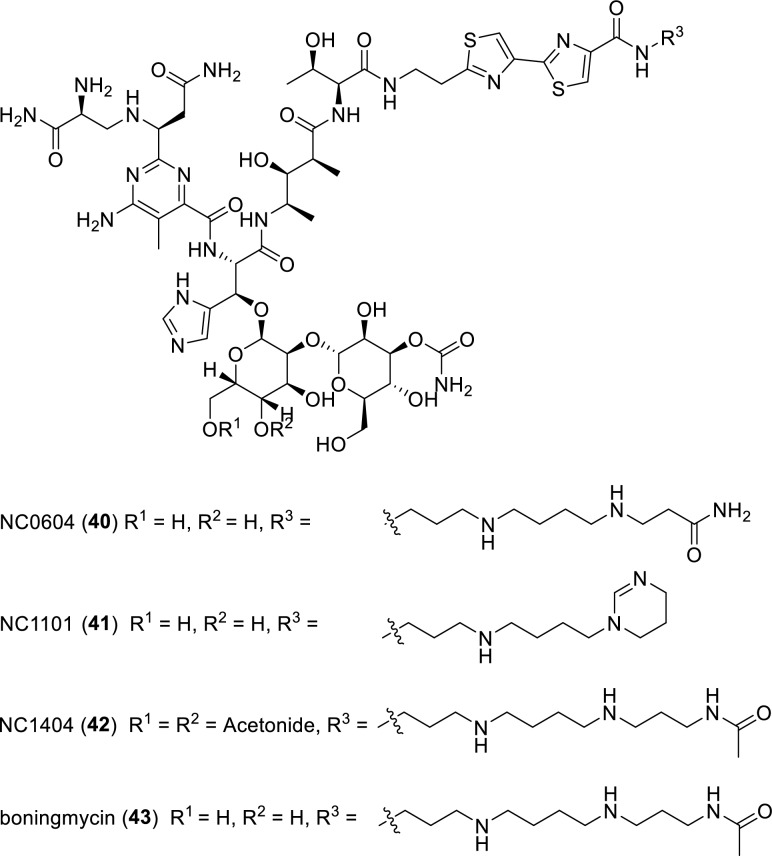 Figure 7
Figure 7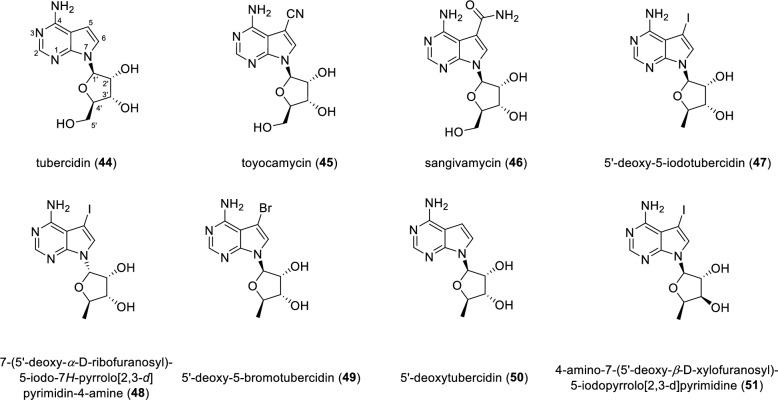 Figure 8
Figure 8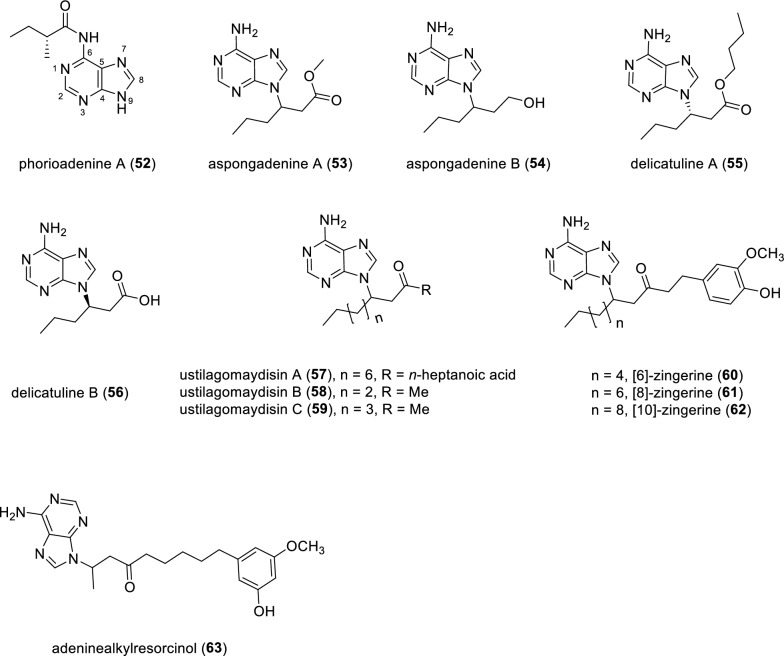 Figure 9
Figure 9 Figure 19
Figure 19- —http://dx.doi.org/10.13039/501100001809National Natural Science Foundation of China
- —Yunnan Fundamental Research Projects
- —Yunnan Fundamental Research Kunming Medical University Projects
- —Open Research Fund of Yunnan Provincial Key Laboratory of Pharmacology for Natural Products
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsBiochemical and Molecular Research · Synthesis and Reactivity of Heterocycles · Multicomponent Synthesis of Heterocycles
Introduction
Natural products have consistently played an integral role in drug discovery and development, profoundly safeguarding human health through a multitude of therapeutic applications [1–3]. Irrefutably, a significant proportion of these natural occurrences have been used directly as active pharmaceutical ingredients or serve as chemical templates for further structural modification into new chemical entities. Moreover, natural products feature rich sp^3^-hybridzed carbons and novel skeletons, which extremely expand molecular diversity and enrich chemical space. These merits contribute to leverage natural product-based drug discovery spanning various medical treatments for diseases, including cancer, cardiovascular diseases, infectious diseases and central nervous system disorders. Additionally, natural products represent a valuable source of chemical probes for chemoproteomic studies designed to target identification and elucidating biological mechanisms [4, 5].
The discovery and development of therapeutics derived from natural products constitutes a multifaceted process involving multiple critical stages, each playing an indispensable role in translating the potential of isolated compounds into viable clinical drugs (Fig. 1).Fig. 1. Key stages in the drug development pipeline derived from natural products
Nitrogen heterocycles are widely distributed among approved drugs [6]. Pyrimidine-based heterocycles, for example, rank as the second most common ring systems in marketed pharmaceuticals (Fig. 2). Pyrimidine and its derivatives have been recognized as privileged scaffolds for drug development in recent years [7–11]. Several approved small-molecule drugs have showcased that pyrimidine ring system is essential to potent biological activities (Fig. 3), including anti-infective, antibacterial, antihypertensive, antiviral, antiplatelet and anticancer effects. Novel pyrimidine-based drug candidates have lured considerable attention, presumably due to its inherent capacity to enhance the binding affinity via hydrogen bond and/or to function as bioisosteres of the phenyl ring and other π-conjugated scaffolds in drug design [12]. Despite the enormous value of this pyrimidine core, comprehensive reviews on natural products that contain substituted or ring-fused pyrimidine ring systems remain relatively scarce compared with those on small-molecule compounds driven by synthetic approaches [13, 14].Fig. 2. Top 10 nitrogen heterocycles in U.S. FDA approved drugs (January 2013–December 2023)Fig. 3. Chemical structures of marketed drugs containing the pyrimidine scaffold
Pyrimidine is an aromatic six-membered heterocyclic compound that consists of four carbon atoms and two nitrogen atoms located at positions 1 and 3. The name “pyrimidine” was first coined by the organic chemist Pinner in the early 1880s. Pinner also pointed out the structural similarity between pyrimidine and other aromatic compounds such as benzene and pyridine, underscoring the importance of its aromatization [15, 16]. It’s noteworthy that pyrimidine-containing natural products and their synthetic analogues continue to serve as irreplaceable engines for therapeutic innovation and aspiration, consistently affording structurally bioactive architectures, including meridianins A-H [17], cordycepin [18] and aplithianine A [19, 20]. Driven by long-standing interest in small-molecule compounds featuring the pyrimidine motif [21–26], this review offers a comprehensive structural profile of pyrimidine-containing natural products, along with an overview of their biological activities, covering the literature from 2004 to early 2025. Furthermore, this review mainly deals with natural products bearing aromatic heterocyclic ring system pyrimidine, while those natural products merging with nonaromatic pyrimidine derivatives, such as 2-pyrimidone, 4-pyrimidone, barbituric acid or three types of nucleobases (cytosine, thymine and uracil) are excluded.
In this review, 156 naturally occurring compounds containing pyrimidine-based motif have been reported, demonstrating a significant variety of chemical structures. The compounds in this review are categorized into three classes, including those with non-glycosylated pyrimidine-containing natural products, glycosylated pyrimidine derivatives and purine-containing natural products.
Notably, two seminal reviews compiled by Lagojia and Rosemeyer respectively have disclosed that the pyrimidine-containing natural products reported before 2004 [27, 28]. However, several pyrimidine-containing structures were unintentionally omitted in their reviews. Given the structural diversity of these natural products discussed and the significance of the topics highlighted, the missing molecules in previous reviews are included in this review.
Non-glycosylated pyrimidine-containing natural products
Non-fused pyrimidine-containing alkaloids
Pyrimidine-containing alkaloids represent a class of natural products characterized by the presence of one or more non-fused substituents on the pyrimidine scaffold. Common examples of such peripheral modifications include indole rings and benzyl groups.
A monosubstituted pyrimidine derivative, 2-(4-pyrimidinyl)-1H-benzimidazole (1), was recently isolated from cultured mycelia of Ophiocordyceps sinensis, which is a conspicuous traditional medicine (Fig. 4). Its chemical structure was established on the basis of mass spectroscopic data analysis [29]. Previous literature indicates that compound 1 was first obtained via chemical synthesis as early as 1960 [30]. Compounds 2–5 are a new class of alkaloids featuring a pyrimidine-β-carboline moiety in their chemical structures. Ingenine A (2), annomontine (3) and ingenines C and D (4 and 5) were isolated from the Indonesian sponge Acanthostrongylophora ingens by Ibrahim and co-workers [31, 32]. Interestingly, the co-isolated alkaloids 2 and 3 showed significant discrepancies in their NMR chemical shifts around the pyrimidine motif and in their optical rotation (OR) values with specific rotations of \documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$$[\alpha]_{\mathrm{D}}^{25} $$\end{document} + 16 for 2 and \documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$$[\alpha]_{\mathrm{D}}^{25} $$\end{document} -34 for 3. These observations suggest that compounds 2 and 3 may constitute a pair of atropisomers. However, the absolute configurations of both compounds (2 and 3) remain unresolved. Compound 3 exhibited pronounced cytotoxicity against the murine lymphoma L5178Y cancer cell line with ED_50_ (median effective dose) value of 7.8 μg·mL^−1^. Compounds 4 and 5 showed moderate inhibitory activity against MCF-7 and HCT-116 cell lines with IC_50_ (half maximal inhibitory concentration) values of 4.33 and 6.05 for 4, and 2.90 and 3.35 μM for 5, respectively. A novel antileishmanial pyrimidine-β-carboline alkaloid, N-hydroxyannomontine (6), was isolated from the bark of Annona foetida via bioassay-guided fractionation [33]. Compound 6 plays a key role in Annona genus chemotaxonomy. From the Indonesian sponge Acanthostrongylophora ingens, ingenine B (7), which features a pyrimidine-γ-carboline motif, was isolated and demonstrated cytotoxicity against lymphoma L5178Y cancer cell line with ED_50_ value of 9.1 μg·mL^−1^ [34]. Another metabolite, acanthomine A (8), also obtained from the sponge A. ingens, represents a new type of pyrimidine-dihyro-β-carboline alkaloid [34]. Furthermore, X-ray crystallographic analysis suggested that acanthomine A (8) seems to occur naturally as a racemate [35]. Hyrtinadine A (9), a bis-indole alkaloid with a para-substituted pyrimidine moiety, was isolated from the sponge Hyrtios sp. Compound 9 showed cytotoxicity against murine leukemia L1210 and human epidermoid carcinoma KB cell lines with IC_50_ values of 1 μg·mL^−1^ and 3 μg·mL^−1^, respectively [36, 37]. From the sponge Scalarispongia sp., an indole-pyrimidine alkaloid hyrtinadine B (10) was isolated and further evaluated its cytotoxicity against the human leukemia cell K562 cell line. Compound 10 showed negligible cytotoxicity [38].Fig. 4. Chemical structures of compounds 1–18
The marine natural products meridianins A-G, characterized by indole-pyrimidine ring systems, represent a new family of protein kinase inhibitors [39, 40]. These alkaloids were purified from the South Atlantic Ascidian Aplidium meridianum. Since their discovery, continuous efforts have been devoted to exploring their biological activities in-depth and developing efficient chemical synthesis routes [41–43]. Recently, a new member of this family, meridianin H (11), along with four structurally related derivatives coupled with 2-aminopyrimidine ring australindolones A–D (12–15) were isolated from the deep water Antarctic tunicate Synoicum sp. [44].
Axistatins 1**–**3 (16–18) with pyrimidine diterpene scaffolds were identified from the Republic of Palau marine sponge Agelas axifera. Compounds 16–18 demonstrated moderate inhibitory activity against cancer cell growth and also exhibited antimicrobial activity [45].
Fused pyrimidine alkaloids
Fused pyrimidine alkaloids are characterized by the presence of two or more cyclic rings in the molecules (Fig. 5). Crucially, at least one of these rings is fused directly to the pyrimidine ring, forming new fused-ring frameworks. A total of 21 such compounds have been identified. Marine-derived mirabilins A–C (19–21) were isolated from the sponge Arenochalina mirabilis, collected from the Great Australian Bight [46, 47]. Netamines F–J (22–26) coupled with aromatic pyrimidine moieties were identified from the marine sponge Biemna laboutei. Interestingly, hydrogenation of netamine H over Pd/C with H_2_ afforded natamine A, suggesting that the relative configuration of the side chains embedded in the molecules is indeed identical. Biological evaluation results indicate that netamine K and mirabilin A exhibited antimalarial activity with IC_50_ values of 2.4 and 20.7 μM, respectively [48]. Two tricyclic alkaloids, 1,8a;8b,3a-didehydro-8β-hydroxyptilocaulin (27), and 1,8a;8b,3a-didehydro-8α-hydroxyptilocaulin (28) were uncovered from the marine sponge Monanchora unguifera. X-ray crystallographic analysis of co-crystallized compounds 27 and 28 confirmed these two compounds as a pair of C-8 epimers [49]. Co-existing compounds 27 and 28 showed antimalarial activity against the parasite Plasmodium falciparum, with an IC_50_ value of 3.8 μg·mL^−1^. Arbusculidine A (29) represents the first compound in the netamine family to feature a benzyl group, whose chemical structure is closely related to the family of tricyclic pyrimidine alkaloids [50]. From a broader perspective, compound 29 also belongs to the distinctive quinazoline skeleton.Fig. 5. Chemical structures of compounds 19–34
Chemical investigation of the Indonesian marine sponge Stylissa carteri led to the isolation of two aminopyrimidine-containing alkaloids latonduines A and B (30 and 31). These two alkaloids with unprecedented tricyclic scaffolds were established by analysis of HRMS and NMR data and further confirmed by the total synthesis of compound 30. Biosynthetically, amino acid ornithine likely serves as the biosynthetic precursor to the aminopyrimidine moiety of latonduines A and B. Both compounds 30 and 31 were evaluated for their in vitro cytotoxicities against a panel of human cancer cell lines and for inhibitory activities against a panel of protein kinases. Unfortunately, they were found to be inactive in these bioassays [51]. Two pyrrole-pyrimidine alkaloids, 3-debromolatonduine A (32) and 3-debromolatonduine B methyl ester (33), were identified from the marine sponge Stylissa sp. [52]. Millaurine A (34), a pyrimidine-containing alkaloid with 6/5/6 tricyclic skeleton, was isolated from the seeds of Millettia laurentii [53].
Investigation of the traditional medical herb Peganum harmala led to the isolation and identification of one quinazoline alkaloid pegaharmaline B (35) (Fig. 6). Notably, compound 35 represents a β-carboline-quinazoline hybrid with a highly conjugated C_24_ scaffold [54]. Compound 39 showed cytotoxic activity against HL-60 cell lines with an IC_50_ value of 16.6 μM. A family of unusual depsipeptide-polyketide compounds, known as jasplakinolide, has showed a significant cytotoxicity against renal, prostate and CNS tumor cell lines with good selectivity. These impressive data stimulate medicinal chemists to uncover more siblings in this family. Finally, jasplakinolide T (36), a new member characterized with quinazoline motif in this family, was isolated from marine sponges Aulettas sp. and Jaspis splenedens. The sponge materials from these two different species were combined together by the authors and subsequently subjected to isolation [55]. Unfortunately, the amount of isolated compound 36 was too limited to conduct a bioassay. An active metabolite 37, possessing a quinazoline moiety, was isolated from the Paenibacillus kribbensis. The isolate 37 has strong broad-spectrum antifungal activity [56]. A pair of enantiomers 38 and 39 featuring quinazoline moieties, were isolated from the traditional Chinese medicine Corydalis yanhusuo and their absolute configurations were elucidated by electronic circular dichroism (ECD) calculations. Both compounds 38 and 39 showed acetylcholinesterase (AChE) inhibitory activity in a dose-dependent manner with the IC_50_ values of 14.07 and 13.75 μM, respectively [57].Fig. 6. Chemical structures of compounds 35–39
Pyrimidine-containing glycopeptides
Pyrimidine-containing glycopeptides, as defined in this review, refer to glycopeptides in which a pyrimidine moiety is incorporated into the molecular framework. Although these compounds include sugar residues, the sugar units are not directly linked to the pyrimidine core. Therefore, they are not classified as glycosylated pyrimidines in strict sense.
Four glycopeptide antibiotics, NC0604 (40), NC1101 (41), NC1404 (42) and boningmycin (43), which contain with tetrasubstituted pyrimidine moiety, are members of the bleomycin family (Fig. 7). Two representative members of this family, bleomycin A2 and bleomycin B2, mainly constitute commercial bleomycin, a clinically used anticancer drug [58]. Compounds 40–42 and 43 were isolated from the fermentation broths of Streptomyces verticillus var. pingyangensis n. var and Streptomyces verticillus var. pingyangensis n. sp., respectively [59–62]. Significantly, compared to bleomycin, compound 40 exhibited 3- to 9-fold greater antitumor activity than bleomycin against human HepG2, KB, MCF-7, HCT-116, BCG-813 and MCF-7 cell lines in vitro, while demonstrating substantially lower pulmonary toxicity. In parallel, compounds 41 and 42 also exhibited more potent inhibition against several tumor cell lines than bleomycin. Compound 43 demonstrated strong anticancer activity both in vitro and in vivo, mediated through the induction of apoptosis and cellular senescence, alongside with negligible pulmonary toxicity. A recent report reveals that PD-L1 upregulation in bleomycin-induced senescence requires activation of JAK/STAT signal pathway. These findings establish the groundwork for future studies aimed at elucidating the mechanisms of PD-L1 regulation during cellular senescence [63–65]. From a structural viewpoint, these four anticancer antibiotics 40–43 share the same core scaffold as clinical drug bleomycin, but feature distinct bithiazole tails that facilitate linkage with long-chain modifications. Therefore, further in-depth research on the structure–activity relationships (SAR), mode of action and toxicity profile of these four compounds is highly promising.Fig. 7. Chemical structures of compounds 40–43
Glycosylated pyrimidine derivatives
Glycosylated pyrrolopyrimidine derivatives constitute a subclass of natural products in which a sugar is directly linked to the pyrrolopyrimidine core. A N-glycosylated fused pyrimidine derivative (Fig. 8), tubercidin (44), was first isolated from Streptomyces tubercidicus. Tubercidin represents a distinctive class of pyrimidine derivatives featuring a pyrrolo[2,3-d]pyrimidine scaffold. This pyrrolopyrimidine nucleoside 44 exhibits broad biological activity, including notable antiviral and anticancer properties. Tubercidin has been shown to strongly inhibit the growth of Streptococcus faecalis (8043), with an IC_50_ value of 0.02 μM. Moreover, compound 44 displayed pronounced cytotoxicity against human cancer lines A549 and HST116, with IC_50_ values of 0.044 μM and 0.043 μM, respectively [66, 67]. In addition, compound 44 was recently identified as a potent methyltransferase 1(MTr1) inhibitor capable of suppressing influenza A replication [68]. Another tubercidin congener, toyocamycin (45), which bears a cyano group at C-5, was isolated from Streptomyces toyocaensis. Subsequent studies revealed that unamycin B, E-212 and vengicide isolated from Streptomyces fungicidicus, Streptomyces sp*.* E-212 and Streptomyces vendargensis respectively were structurally identical to toyocamycin [69, 70]. Compound 45 is a specific CDK9 inhibitor with an IC_50_ value of 79 nM. Compound 45 also inhibited XBP1 mRNA splicing with an IC_50_ value of 80 nM and suppressed tumor growth in a xenograft model of human multiple myeloma [71]. Sangivamycin (46) was first isolated from an unidentified Streptomyces species and later also rediscovered from the fermentation broth of Streptomyces rimosus. Compound 46 inhibited protein kinase C (PKC) with a Ki value of 10 μM and showed potent antiproliferative activity against HFF human fibroblasts, with an IC_50_ value of 0.08 μM [72, 73].Fig. 8. Chemical structures of compounds 44–51
A pair of C1' anomers, 5'-deoxy-5-iodotubercidin (47) and 7-(5'-deoxy-α-D-ribofuranosyl)-5-iodo-7H-pyrrolo[2,3-d] pyrimidin-4-amine (48), were isolated from the red alga Hypnea valendiae [74]. Two pyrrolopyrimidine nucleosides, 5'-deoxy-5-bromotubercidin (49) and 5'-deoxytubercidin (50) were obtained as naturally occurring compounds from the ascidian Didemnum voeltzkowi [75]. Notably, these two tubercidin analogs (49 and 50) have previously prepared by semi-synthesized prior to their discovery as natural products [76]. Among them, compound 49 demonstrated potent inhibition of adenosine kinase with an IC_50_ value of 0.04 μM [77]. An unusual 5'-deoxyxylofuranosyl nucleoside, 4-amino-7-(5'-deoxy-β-D-xylofuranosyl)-5-iodopyrrolo[2,3-d]pyrimidine (51), was isolated from the ascidian Diplosoma sp. collected along the coast of Hateruma island, Okinawa [78]. Total synthesis of compound 51 was subsequently accomplished from the D-xylose, enabling unambiguous establishment of its absolute configuration [79].
Purine-containing natural products
The heterocyclic aromatic organic compound purine consists of a pyrimidine ring fused to an imidazole ring. Purine derivatives constitute a wide variety of natural products containing pyrimidine ring system. Previous pioneering review covered by Rosemeyer provided a comprehensive summary about purine-containing natural products described before 2004 [28]. And several other reviews closely related to purine ring system highlight terpenyl-purine natural products and synthesis of natural purine nucleoside antibiotics [13, 14]. In this review section, natural products incorporating aromatic purine ring system will be exhaustively summarized.
Purine alkaloids
Purine alkaloids constitute a diverse class of purine derivatives, encompassing N-alkylated purine alkaloids, terpenylated purines and miscellaneous metabolites characterized by an aromatic purine moiety. The* N*-alkylated subgroup includes compounds in which a purine nitrogen is substituted by alkyl chain. Terpenylated purines comprise conjugates in which the purine nucleus is fused or appended to sesquiterpenoid, diterpernoid, steroid and triterpenoid fragments. Compounds that do not fall into these categories are classified herein as miscellaneous purine derivatives. In addition, two terpenylated purine alkaloids (compounds 99 and 102), in which a sugar unit is indirectly tethered to the purine core, are included in this section. In parallel, two purine alkaloids (compounds 101 and 103) simultaneously bearing a sugar part and N-glycosylated purine are also compiled in this section, highlighting the importance of steroid and triterpenoid in this subgroup.
N-Alkylated purines
Chemical investigation of the southern Australian marine sponge Phoriospongia sp. (CMB-03107) afforded a naturally occurring alkaloid phorioadenine A (52), representing the first example of a 6-N-acyladeine (Fig. 9). The structure of compound 52 was elucidated by spectroscopic analysis and further confirmed by the chemical synthesis of racemic and enantiomeric forms of 52 [80]. Of note, rac-52, ent-52 and three analogues (with differences located at the N-acyl side chains) were inactive toward the inhibition of larval development of Haomonchus contortus. Only compound 52 exhibited nematocidal activity against H. contortus with the LD_99_ value of 31 μg·mL^−1^, indicating the importance of the nature and chirality of the N-acyl side chain. Interestingly, the author conjured that compound 118 was likely to be a co-isolate, and subsequently completed the total synthesis of compound 118 (see Fig. 15). HPLC-ESIMS data analysis of authentic crude extract of Phoriospongia sp. (CMB-03107) successfully detected phorioadenosine A (118) as a minor co-existed secondary metabolite. It appears that the amount of compound 118 was too limited to be isolated and fully characterized.Fig. 9. Chemical structures of compounds 52–63
Two adenine analogues, aspongadenines A and B (53 and 54), were naturally isolated as racemates from the insect Aspongopus chinensis. Chiral HPLC separation of these two racemic compounds 53 and 54 afforded two pairs of enantiomers, respectively. The absolute configurations of these optically pure compounds (R)-53 and (S)-53 were determined by calculated ECD. Subsequently, the absolute configurations of these optically pure compounds (R)-54 and (S)-54 were tentatively assigned by comparison of OR values with those of the enantiomers of compound 53 [81]. Two adenine-containing compounds delicatulines A and B (55 and 56) were isolated from the n-butanol extract of Selaginella delicatula. The absolute configurations of compounds 55 and 56 were determined by comparing their experimental ECD spectra with those of reported asponguanine A. Compounds 55 and 56 showed no inhibitory effect on HBV in the antiviral activity assay [82].
Ustilagomaydisins A–C (57–59), three adenine-derived alkaloids, were obtained from the pathogenic fungus Ustilago maydis. The OR values of the isolated compounds (57–59) were nearly zero, suggesting that these three isolates could be racemic. Compounds 57 showed weak activity to reverse the multidrug resistant in doxorubicin-resistant K562/A02 cell lines with verapamil as a positive control [83]. Phytochemical study on the methanol extract of ginger rhizomes (Zingiber officinale) led to the isolation and structure elucidation of three adenine derivatives [6]-,[8]-, and [10]-zingerines (60–62) as racemates. It’s noteworthy to point out that chemical structures of these three compounds (60–62) were further confirmed by chemical synthesis. A Michael addition between [6]-shogaol and adenine gave the desired molecule 60. Using [8]-shogaol and [10]-shogaol under the identical reaction conditions, compounds 61 and 62 were smoothly prepared, respectively [84]. Adeninealkylresorcinol (63) is an alkylresorcinol derivative isolated from Lasiodiplodia sp., bearing an adenine moiety [85].
Terpenylated purines
An adenine-11β,13-dihydrolectuca hybrid (Fig. 10), named 11β,13-(6-amino-purine-9-yl)-dihydrolectuca (64), was unlocked from Lactuca tatarica. Compound 64 displayed good inhibitory activity on Bruton’s tyrosine kinase with an IC_50_ value of 2.75 μM and also anti-inflammatory activitiy against NO release from mouse RAW264.7 macrophages induced by LPS with an IC_50_ value of 7.09 μM [86]. Pestalotiphain A (65), the first example of a β-caryophyllene derivative containing an adenine moiety, was identified from a plant-associate fungus Pestalotiopsis hainanensis. Compound 65 was originally described as a pair of hemiacetal epimers at C-14 (2.5:1 ratio in methanol-d^4^) based on a comprehensive analysis of NMR data. The absolute configuration of compound 65 was assigned by comparing its experimental ECD and OR value with those of the co-isolated pestalotiopsin C, a β-caryophyllene-type sesquiterpene with methoxy group at C-7 [87]. Aneudesmanolide-type sesquiterpenoid (66), combined with an adenine moiety, was characterized from the flower heads of Sphaeranthus indicus. In addition, semisynthesis of compound 66 was achieved through a Michael addition reaction of the natural isolate 7-hydroxyfrullanoide and adenine, further rationalizing its chemical structure [88].Fig. 10. Chemical structures of compounds 64–66
Five adenine-diterpene secondary metabolites, asmarines G–K (67–71), were isolated from the marine sponge Raspailia sp. (Fig. 11). All five isolated compounds (67–71) were evaluated for their in vitro cytotoxicities against a panel of human cancer cell lines. However, they were found to be inactive, in stark contrast to the potent congener asmarine A [89, 90].Fig. 11. Chemical structures of compounds 67–71
The genus Agelas, originating from the marine sponge, has been recognized as a rich source of diterpenes coupled with poplar functionality adenine [91]. Three diterpene alkaloids (Fig. 12), agelasines J–L (72–74), which belong to the agelasine family and possess a 9-N-methyladeninium, were isolated from the marine sponge Agelas sp*.* [92]. These three compounds (72–74) exhibited mild antimalarial activities against Plasmodium falciparum with IC_50_ values of 6.6, 8.3 and 18 μM, respectively. Another sibling agelasine M (75) were uncovered from the sponge Agelas sp. collected in Papua New Guinea [93]. A follow-up bioassay revealed that compound 75 showed notable antiparasitic activity against Trypanosoma brucei with an IC_50_ value of 3.0 μg·mL^−1^, alongside less cytotoxicity against Jurkat cell T. brucei.Fig. 12. Chemical structures of compounds 72–86
The diterpene alkaloid agelasine N (76), was isolated from the Caribbean sponge Agelas citrina. The relative configuration of compound 76 was assigned by NOE correlation. However, the absolute configuration of compound 76 remains elusive due to the limited availability of the sample [94]. Agelasines O–U (77–83), possessing three different diterpene skeletons tethered with 9-N-methanyladenine motifs, were identified from the marine sponge Agelas sp. collected at Okinawa [95]. Conspicuous compounds (77–80) are endowed with the pyrrole esters and show antimicrobial activities against several bacteria and fungi. Three diterpene alkaloids containing an N-methyladenine moiety, 2-oxoagelasines A (84) and F (85) and 10-hydro-9-hydroxyagelasine F (86), were isolated from the Okinawan marine sponge Agelas nakamurai. Compound 84 inhibited the growth of Mycobaterium smegmatis with inhibition zones of 10 mm at 20 μg/disc [96].
A congener of agelasine isoagelasine C (87), was isolated from the South China Sea sponge Agelas nakamurai, and its absolute configuration was deduced by ECD calculations (Fig. 13). Compound 87 displayed moderate antibacterial activities against Proteusbacillus vulgaris with MIC value of 18.75 μg·mL^−1^ [97]. Three adenine-containing alkaloids nakamusines A–C (88–90) were isolated from the Taiwanese sponge Agelas nakamurai. The absolute configuration of compound 88 was rationalized by ECD calculations [98]. Interestingly, co-occurring natural product 9-methyladenine was also isolated at the same time, indicating it might serve as a crucial biosynthetic building block to construct adenine-containing diterpene alkaloids in Agelas sponge. Two unique norditerpenoid alkaloids embedded with adenine moiety, gelasines A and B (91 and 92), were isolated from the marine sponge Agelas. sp. Compounds 91 and 92 were inactive in the antiparasitic assay against T. brucei [93]. Nemoechines F and G (93 and 94), featuring N-methyladenine-containing diterpene alkaloids, were isolated from the South China Sea sponge Agelas aff. nemoechinata. The absolute configuration of compound 93 was tentatively assigned on the basis of ECD calculations. Compound 94 showed weak activity against human lymphoblastic leukemia Jurkat cell lines with an IC_50_ value of 17.1 μmol·L^−1^. In the follow-up antimicrobial bioassay against Staphyloccocus aureus and Bibrio parahemolyticus, both compounds were inactive [99].Fig. 13. Chemical structures of compounds 87–98
Two adenine-containing diterpene alkaloids (−)-agelasine D (95) and its oxime derivative (−)-ageloxime D (96) were isolated from the Indonesian sponge Agelas nakamurai. The ^1^H and ^13^C NMR data of compound 95 were identical to those of (+)-agelasine D, whose absolute configuration was established by chemical synthesis [100]. Interestingly, the experimental specific rotation value \documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$$[\alpha]_{\mathrm{D}}^{25} $$\end{document} of compound 95 is −19 (c 0.5, MeOH) while the previously recorded specific rotation value \documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$$[\alpha]_{\mathrm{D}}^{25} $$\end{document} of (+)-agelasine D is + 10.4 ((c 1.1, MeOH), suggesting that the compound 95 is the levorotatory enantiomer of ( +)-agelasine D. Compound 96 was also determined to be levorotatory with the similar method. Compounds 95 and 96 displayed cytotoxicity against L5178Y mouse lymphoma cell line with IC_50_ values of 4.03 and 12.5 μM, respectively. Furthermore, in an anti-fouling bioassay, both compounds 95 and 96 inhibited settling of larvae of Balanus improvius through their cytotoxicities. Ageloxime B (97) was isolated from the marine sponge Agela mauritiana. Compound 97 showed antifungal activity against Cryptococcus neoformans, antileishmanial activity against Leishmania donovani in vitro and antibacterial activity against Staphylococcus aureus and methicillin-resistant Staphylococcus aureus in vitro [101]. An ongoing exploration of the same sponge Agela mauritiana led to isolation of a diterpene alkaloid ( +)-agelasine B (98). The structure of compound 98 was determined as the dextrorotary isomer of agelasine B based on opposite optical rotation data for compound 98 ( \documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$$[\alpha]_{\mathrm{D}}^{25} $$\end{document} + 22.7 MeOH) and for (-)-agelasine B ( \documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$$[\alpha]_{\mathrm{D}}^{25} $$\end{document} -21.5, MeOH). Compound 98 not only showed moderate cytotoxicity against the cancer cell lines PC9, A549, HepG2, MCF-7 and U937 with IC_50_ values of ranging from 4.49 to 14.07 μM, but also displayed antibacterial activities against a panel of methicillin-resistant Staphylococcus aureus (MRSA) strains with MIC_50_ values of 1–2 μg·mL^−1^ [102].
An adenine-containing compound 3β-purine**-**daturmetelide U (99) was isolated from the leaves of Datura stramonium (Fig. 14). Compound 99 showed excellent anti-inflammatory activity against BV2 microglia induced by lipopolysaccharides [103]. An unusual withanolide derivative 3β-(adenin-9-yl)-2,3-dihydrowithaferin A (100) possessing an adenine moiety was isolated from aeroponically grown Withania somnifera, an important herb known as Indian ginseng in Ayurvedic medicine [104, 105]. Cucurbitaglycosides A and B (101 and 102), the first cucurbitane triterpenoids tethered with a purine unit, were discovered from the fruits of Cucurbita pepo cv. dayangua. The relative configurations of compounds 101 and 102 at C-24 remain elusive. Compounds 101 and 102 displayed cytotoxic activity against the Hela cell line with IC_50_ values of 17.2 and 28.4 μg·mL^−1^ [106].Fig. 14. Chemical structures of compounds 99–103
A unique cucurbitane triterpenoid coupled with an adenine motif, machilaminoside B (103), was isolated from the stem bark of Machilus yaoshansis. Despite high degree of structure similarity between compound 101 and compound 103, confirming their identify as the same compound remains challenging. That is because they were obtained from different plant species, and their NMR data were recorded in different deuterated solvents. From another perspective, compounds 101 and 103 could be also classified as N-glycosylated nucleosides (vide infra). Compound 103 exhibited cytotoxic activities against five human cancer cell lines with IC_50_ values ranging from 0.3 to 0.8 μM without selectivity. Additionally, compound 103 also showed TNF-α secretion inhibitory activities in mouse peritoneal macrophages with an IC_50_ value of 0.1 μM [107].
Miscellaneous purine derivatives
A purine derivative, 6-acetylpurine (104) was isolated from culture broth of Cordyceps militaris (Fig. 15). Notably, the OR value of compound 104 was −53.3 recorded in methanol while the compound 104 lacks an apparent chiral center [108]. However, no further explanation was provided by the author for this result. Adenine-9-methylaldehyde oxime B (105), an adenine-containing alkaloid with quite rare oxime unit [109], was discovered from the adult insect of Allomyrina dichotoma. Compound 105 showed inhibitory activities against the Gram-negative bacteria Escherichia coli with diameter of inhibition zone at 5.5 mm [110]. A high-throughput screen performed on approximately 326,000 prefractionated natural product mixtures generated by the National Cancer Institute’s (NCI) Program for Natural Product Discovery (NPNPD), leading to discovery a novel kinase inhibitor, namely, aplithianine A (106) isolated from the marine tunicate Aplidium sp. Unprecedent compound 106 showed potent inhibition against J-PKAcα and wild-type PKA with IC_50_ values of approximately 1 μM and 84 nM. Follow-up mechanistic studies indicated that compound 106 inhibited PKAcα catalytic activity by competitively binding to the ATP pocket. Additionally, concise total synthesis of compound 106 was conquered in 4 steps [19, 20].Fig. 15. Chemical structures of compounds 104–112
Aplidiopsamine A (107), the first tricyclic alkaloid possessing the 3H-pyrrolo[2,3-c]quinoline linkage with an adenine moiety, was identified from the Australian ascidian Aplidiopsis confluata. Compound 107 demonstrated inhibition against two strains of plasmodium falciparum, including chloroquine sensitive (3D7) and resistant strains (Dd2) strains with IC_50_ values of 1.47 μM and 1.65 μM, respectively. More importantly, Compound 107 exhibited minimal toxicity toward human cell line HEK-293 [111, 112]. Alachalasins F and G (108 and 109) represent two unprecedented cytochalasins featuring adenine moieties, which were isolated from the fungus Stachybotrys charatum. Compound 108 showed weak antifungal activity against Staphylococcus aureus [113]. The isolation and characterization of a unique bromophenol derivative (110), coupled with an adenine moiety, from the red alga Rhodomela confervoides was described. Compound 110 represents the first example of bromophenols tethered with adenine moiety with the name 9-[3-bromo-2-(2,3-dibromo-4,5-dihydroxybenzyl)-4,5-dihydroxybenzyl]adenine [114].
Two insect-derived adducts aspongdopamines A and B (111 and 112) were identified from Aspongopus chinensis, bearing unprecedented hybrids consisting of N-acetyldopamine and adenine. Notably, both compounds 111 and 112 were naturally isolated as racemates. Separation via chiral HPLC afforded eight diasteroisomers 111a-111d and 112a-112d, respectively, due to the fixed trans-relationship of H-7' and H-8'. The absolute configurations of these stereoisomers were unambiguously determined by 14-steps total synthesis, ECD and VCD calculations [115]. Compounds 113 and 114, namely**,** ( ±)-4-(6-amino-9H-purin-9-yl)-5,8-dihydroxy-3,4-dihydronaphthalen-1(2H)-one and ( ±)-4-(6-amino-9H-purin-9-yl)-5-hydroxy-3,4-dihydronaphthalen-1(2H)-one, respectively, possess the phenol-adenine hybrids originally isolated as scalemic mixtures from the flowers of Juglans regia. Separation of compound 113 and 114 via chiral HPLC afforded two pairs of enantiomers, respectively. Accordingly, the absolute configurations of these optically pure compounds (R)-113 and (S)-113 and (R)-114 and (S)-114 were determined by ECD data, respectively [116].
N-glycosylated nucleosides
N-glycosylated nucleosides are defined as a class of molecular structures consisting of a base subunit adenine and a sugar subunit. Adenosine, a naturally occurring nucleoside, is one of the building blocks of DNR or RNA. In this section, N-glycosylated alkaloids with aromatic adenine moieties were recapitulated.
Oxetanocin A (115) represents the first naturally occurring alkaloid harboring an oxetanosyl-N-glycoside isolated from the culture broth of Bacillus megaterium NK84-0218 in 1986 (Fig. 16). Due to its interesting antiviral activity against human immunodeficiency virus (HIV), a series of oxetanocin A derivatives, including compound 116, were obtained via chemical modification [117]. Nearly two decades later, compound 116 was described as a natural product isolated from the culture broth of Streptomyces albus subsp. chlorinus NRRL B-24108 via bioassay directed fractionation and was given the trivial name albucidin [118]. Compound 116 showed herbicidal activity against a broad spectrum of weeds. Recently, albucidin and its enantiomeric counterpart were synthesized, highlighting a late-stage reductive deiodination by visible light photocatalysis [119]. X-ray crystallography analysis of synthetic compounds pinpointed the absolute configuration of naturally occurring albucidin as 1R,3S. A very recent study has disclosed that two radical S-adenosyl-L-methionine (SAM) enzymes AlsB and AlsA are responsible for catalyzing a ring contraction step and eliminating a one-carbon fragment respectively, providing new insights into biosynthesis of albucidin [120]. Sorangiadenosine (117) possessing a glycosylated purine alkaloid with a bicyclic eudesmane-type sesquiterpene, was isolated from the culture broth of the myxobacterium Sorangium cellulosum collected from Korean soil. Compound** 117** showed moderate antibacterial activity against Bacillus substilis ATCC 6633 and Micrococcus leuteus IFC 12708 strains, with the MIC values of 6.25 and 6.25 μg·mL^−1^, respectively [121].Fig. 16. Chemical structures of compounds 115–124
Chemical investigation of the n-BuOH-soluble fraction of the marine ascidian Herdmania momus led to the isolation and identification of two glycosylated purine derivatives momusine A (119) and its interconvertible transesterification isomer (120). Notably, compounds 119 and 120 afforded an equilibrium mixture of in a 56:44 ratio after HPLC separation upon standing at room temperature, indicating the presence of non-enzymatic acyl migration, as previously observed in similar precedents [122]. Furthermore, the presence of a chiral sulfoxide group in the compound 119 was ascribed to the co-isolated compound 120. However, the sulfoxide configuration of compound 119 remains unknown. Compounds 121 and 122 are two complex nucleoside derivatives isolated from the ascidian Atriolum robustum. Structurally, compound 121 is quite similar to the compound 122, with the striking differences being the methylthio group in compound 121 and the methylsulfinyl moiety in compound 122. Compound 121 displayed affinity for A_1_ and A_3_ adenosine receptor with Ki values below 10 μM [123]. Xanthones A and B (123 and 124), two adenine-coupled xanthones, were identified from the endophytic ascomycete Chalara sp. 6661. And they were chemically induced by epigenetic manipulation of gene expression [124].
A glycosylated purine derivative, nellielloside A (125), was uncovered from the Pacific bryozoan Nelliella nelliiformis through NMR-guided isolation (Fig. 17). Compound 125 showed potent inhibitory activity against three kinases GSK3A, MAPK14, and RSK2 with IC_50_ values of 0.89, 1.00 and 0.80 μM, respectively [125]. Chemical investigation of the rhizomes of Ligusticum striatum DC. led to the isolation and characterization of two unusual N-10 substituted adenosine alkaloids, liguadenosines A and B (126 and 127) [126]. Although both compounds 126 and 127 showed significant anti-platelet aggregation activities in a concentration-dependent manner, their inhibitory effects were strikingly distinctive. Specifically, compound 126 displayed the strongest inhibitory effect at 10 μM and only weak inhibitory effect at 100 μM, whereas compound 127 showed the strongest inhibitory effect at 100 μM and weak inhibitory effect at 10 μM. The specific reason underlying this unusual phenomenon remains unclear. Four members of puromycin family, puromycins B–E (128–131), containing amino-nucleoside hybrids, were isolated from the Himalayan Streptomyces sp. PU-14-G collected at the Bara Gali region of northern Pakistan [127]. The absolute configuration of amino acid residue in compound 128 was determined to be L-leucine by Marfey’s method. Notably, compound 128 represents the first puromycin-related secondary metabolite with a 3'-N-amino acid substitution, in contrast to the classical 3'-N-tyrosinyl substitution in puromycin-type natural products. Ostrerine A (132), a dimeric alkaloid appended with a glycosylated purine, was isolated from the Quanzhou marine mollusk Ostrea rivularis [128]. A glycosylated purine, N^6^-(4-hydroxybenzyl)-adenine riboside (133), was obtained from the rhizomes of Gastrodia elata, a traditional Chinese medicine [129]. Compound 133 potently prevented PC12 cell apoptosis in a concentration-dependent manner with an IC_50_ value of 4.66 μM.Fig. 17. Chemical structures of compounds 125–133
Four glycosylated purine derivatives (Fig. 18), cordycepin (134), adenosine (135), N^6^-(2-hydroxyethyl)adenosine (136) and 5'-(3''-deoxy-b-D-ribofuranosyl)-3'-deoxyadenosine (137), were identified from Cordyceps militaris, which is less well known to the public compared with Cordyceps sinensis (Dong Chong Xia Cao), a flagship species of the genus widely utilized in traditional medicine across Asian countries [130]. Vantagepoints of cordycepin chemistry continues to enjoy a renaissance, although it has been previously described long ago [131–134]. Another two glycosylated purine derivatives, N^6^-4-methylbutyrat-adenosine (138) and 3'-deoxy-6-O-methylinosine (139), were also disclosed from the fruiting bodies of Cordyceps militaris [135]. Herbicidins constitute a group of adenosine-derived nucleoside natural products, herbicidins A-C (140–142), herbicidins E–H (143–146), herbicidin K (147), aureonuclemycin (148), herbicidins L-N (149–151), 8'-epi-herbicidin F (152), 2'-O-demethylherbicidin F (153), 8'-epi-herbicidin B (154), 9'-deoxy-8',8'-dihydroxyherbicidin B (155) and 9'-deoxy-8'-oxoherbicidin B (156), which were isolated from various Streptomyces strains [136–139]. Structurally, they share a common scaffold decorated with an unusual tricyclic undecose, but differ in their substitution patterns at C2', C8', C9' and C11'-position. Aureonuclemycin (148), however, lacks substitution at these positions and consists solely of the nucleoside core. The characteristic tricyclic core of herbicidins comprises a furano-pyrano-pyran moiety appended with a (5-hydroxy)tiglyl unit [140–142]. The biosynthetic pathway was deciphered via feeding experiments, indicating that the tricyclic core originates from D-glucose and D-ribose and tiglyl unit is derived from an intermediate of L-isoleucine [143, 144].Fig. 18. Chemical structures of compounds 134–156
Discussion and outlook
The past two decades have witnessed a surge in effort to discover and characterize pyrimidine-containing natural products with fascinating structural attributes, such as ring-system complexity, richness in sp^3^-hybridized carbons, heteroatom contents and numerous stereocenters, providing an inspiration for medicinal chemists and chemical biologists to design novel molecules that leverage pyrimidine core as a privileged scaffold. Although the pyrimidine-containing natural occurrences in this review occupy only a limited proportion of total natural product repertoire identified hitherto, their structural diversity holds significant promise for anticancer, antiviral and antibacterial therapeutics. Despite being regarded as promising sources of novel biological activities, the full therapeutic value of these pyrimidine-containing natural products remains largely untapped due to the scarcity of authentic samples from natural sources. In this review, pyrimidine-containing compounds are structurally classified into different subgroups, offering a convenient reference for previously undescribed analogues in the foreseeable future and helping to intuitively renew interest in these underutilized compounds as promising synthetic targets.
Identified pyrimidine-containing natural products show diverse biological activities, including anticancer, antiviral and antibacterial activities. This interesting phenomenon closely mirrors the trends observed in synthetically derived small-molecule compounds. Several kinase inhibitors of pyrimidine-containing natural products, aplithianines A, nellielloside A and variolin B, exhibit potent activities in the nanomolar range and represent unique heterocyclic frameworks with considerable promise for the development of anticancer therapeutic. Tubercidin and their derivatives capable of suppressing the influenza hold potential as antiviral drug leads. However, the translation of these potent natural products into clinically viable drug candidates remains challenging. One promising strategy involves integrating these pyrimidine-containing natural products with rationally designed fragments to generate pseudonatural products, thereby enabling the systematic optimization of lead candidates. This approach may improve target selectivity, reduce cytotoxicity and minimize off-target effects. Moreover, further mechanism of action studies, particularly in animal-based disease models, would be highly beneficial for elucidating their target-specific structure–activity relationships and advancing these compounds towards therapeutic development.
With the advancement of new organic synthetic methodology developed, biosynthetic pathways elucidated and AI-driven cheminformatics integrated towards pyrimidine-containing compounds, daunting challenge posed by the paucity of authentic natural samples will be addressed, which will help unlock the full potential of these pyrimidine-containing compounds. Designing proteolysis-targeting chimaeras (PROTACs) based on pyrimidine-containing natural products offers an emerging option that holds potential to revolutionize contemporary drug discovery. Furthermore, continued efforts in the isolation and biological evaluation of such natural products will lay a solid foundation for future drug discovery.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Newman DJ, Cragg GM. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J Nat Prod. 2020;83:770–803.10.1021/acs.jnatprod.9b 0128532162523 · doi ↗ · pubmed ↗
- 2Butler MS, Capon RJ, Blaskovich MAT, Henderson IR. Natural product-derived compounds in clinical trials and drug approvals. Nat Prod Rep. 2025. 10.1039/D 5NP 00031 A 10.1039/d 5np 00031 a 40923383 · doi ↗ · pubmed ↗
- 3Luo ZW, Yin FC, Wang XB, Kong LY. Progress in approved drugs from natural product resources. Chin J Nat Med. 2024;22:195–211.10.1016/S 1875-5364(24)60582-038553188 · doi ↗ · pubmed ↗
- 4Swati, Shaveta, Shalini, Sharma S, Kumar R, Kumar K, Kumar V. Advances in the synthesis and anticancer activities of pyrimidine based scaffolds. Eur J Med Chem. 2026;302:118319.10.1016/j.ejmech.2025.11831941218520 · doi ↗ · pubmed ↗
- 5Liao M, Shu C, Hu L, Zhao P, Zheng W. Paenibacillus kribbensis metabolite and application thereof to biocontrol. Chinese patent, CN 109265461 A, 2019-1-25.
- 6Liu X, Fu M, Qin Z, Lei G. Extraction of 11β,13-(6-amino-purine-9-yl)-dihydrolectuca from Lactuca tatarica. Chinese patent CN 116621837, 2023-8-22.