Artisanal food of animal origin as reservoir of putative pathogenic Escherichia coli: a combined genomic and in vivo approach
Frédérique Pasquali, Cecilia Crippa, Alex Lucchi, Gerardo Manfreda

TL;DR
This study explores how artisanal foods like cheese and salami may carry harmful Escherichia coli strains, using genomic and in vivo methods to assess their pathogenic potential.
Contribution
The study combines genomic analysis and in vivo testing to identify pathogenic E. coli strains in artisanal foods and their potential sources.
Findings
Raw materials, not processing environments, are the main source of E. coli contamination in salami.
Some E. coli strains from salami are closely related to human and wild boar strains, suggesting potential reservoirs.
Genes like espC and malX are enriched in clinical E. coli genomes and may indicate higher risk to consumers.
Abstract
The lack of a full automation and control of environmental parameters might result in potential risk of microbial contamination in small-scale production plants such as artisanal cheese and salami Italian productions. In a previous study, genomes of 33 E. coli isolates were sequenced. In the present study, the pathogenicity potential of E. coli strains was investigated by: (1) phylogenomic comparison with 202 public genomes of human, animal and environmental Italian origin; (2) pathogenicity assessment of strains with virulence patterns predicting specific E. coli pathotypes by using larvae of Galleria mellonella as in vivo infection model. Phylogenetic reconstruction revealed raw material and not the processing environment as source of salami contamination. Moreover, close proximity of some strains isolated from salami production with wild boar and extraintestinal human public strains…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
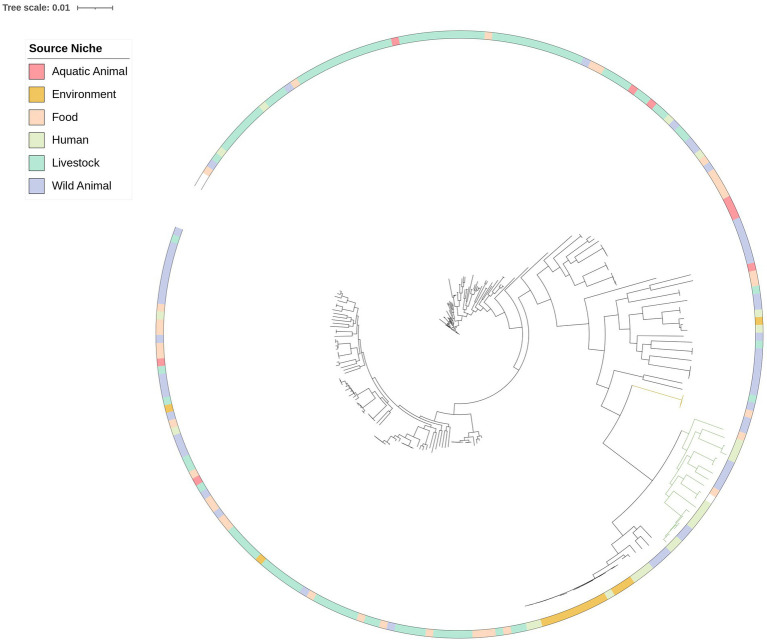 Figure 1
Figure 1 Figure 2
Figure 2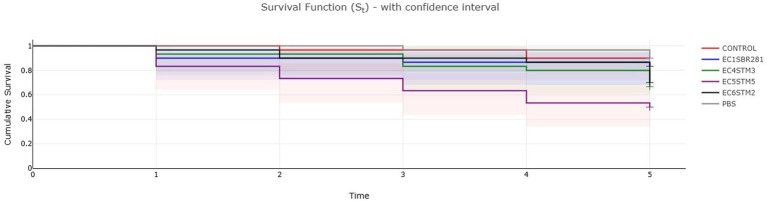 Figure 3
Figure 3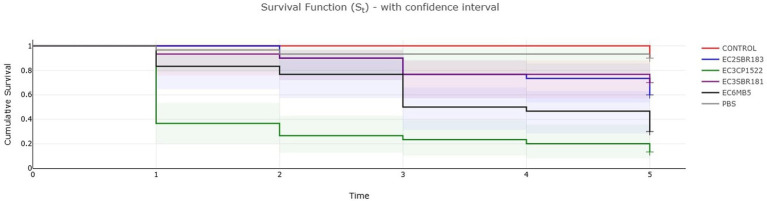 Figure 4
Figure 4| Sample | ANI (%) | Food production | Sample origin | Batch |
|---|---|---|---|---|
| 3SBR3 | 99.73 | Salami | Semi-finished product | 3 |
| 6 MB5 | 99.94 | Salami | Raw material | 6 |
| 5SBR5 | 99.94 | Salami | Semi-finished product | 5 |
| 4 MB1 | 99.88 | Salami | Raw material | 4 |
| 5SBR281 | 98.90 | Salami | Finished product | 5 |
| 6CP4 | 99.92 | Cheese | Finished product | 6 |
| 5SBR101 | 99.82 | Salami | Semi-finished product | 5 |
| 5STM5 | 98.94 | Salami | Environment | 5 |
| 1SBD5 | 98.91 | Salami | Semi-finished product | 1 |
| 6STM2 | 99.80 | Salami | Environment | 6 |
| 3 MB1 | 99.87 | Salami | Raw material | 3 |
| 6SWM3 | 99.83 | Salami | Environment | 6 |
| 3CP1522 | 99.92 | Cheese | Finished product | 3 |
| 2SBR183 | 99.82 | Salami | Semi-finished product | 2 |
| 3SM1 | 99.87 | Salami | Environment | 3 |
| 3SBD4 | 99.85 | Salami | Semi-finished product | 3 |
| 3CW2 | 99.92 | Cheese | Semi-finished product | 3 |
| 1SBR281 | 99.35 | Salami | Finished product | 1 |
| 4SBD3 | 98.73 | Salami | Semi-finished product | 4 |
| 4SBR281 | 99.79 | Salami | Finished product | 4 |
| 4SM3 | 99.71 | Salami | Environment | 4 |
| 4STM3 | 99.46 | Salami | Environment | 4 |
| 5 MB1 | 99.65 | Salami | Raw material | 5 |
| 6SBR282 | 99.83 | Salami | Finished product | 6 |
| 1SBR181 | 98.88 | Salami | Semi-finished product | 1 |
| 2 MB1 | 99.32 | Salami | Raw material | 2 |
| 2SBD5 | 99.91 | Salami | Semi-finished product | 2 |
| 2SBR103 | 99.82 | Salami | Semi-finished product | 2 |
| 2SBR282 | 99.89 | Salami | Finished product | 2 |
| 2SBR4 | 99.35 | Salami | Semi-finished product | 2 |
| 3SBR181 | 99.81 | Salami | Semi-finished product | 3 |
| 1SBR3 | 98.74 | Salami | Semi-finished product | 1 |
| 3SBR281 | 98.90 | Salami | Finished product | 3 |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsEscherichia coli research studies · Vibrio bacteria research studies · Listeria monocytogenes in Food Safety
Introduction
1
In recent years, the demand for more genuine food has been shifting consumer choice toward artisanal foods (Almli et al., 2011; Cirne et al., 2019). Locally produced dairy and meat-based foods such as soft cheese and salami are among foods rising interest of consumers searching for food perceived as healthier (Roccato et al., 2017; Jaramillo-Bedoya et al., 2021). In particular in Italy, already since 2017, 76% of Italian consumers declared to be oriented to national products with specific focus on Protected Designation of Origin (PDO) and Protected Geographical Indication (PGI; Eurispes, 2017). Specifically in the dairy sector, at present 48% of Italy’s cow’s milk supply is dedicated to DOP/IGP cheeses (Assolatte, 2025). However, artisanal foods are produced in small-scale production plants, where the lack of a full automation and control of environmental parameters might result in potential risk of microbial contamination (Roccato et al., 2017; Jaramillo-Bedoya et al., 2021; Pasquali et al., 2023; Pasquali et al., 2022).
Escherichia coli is a facultative, anaerobic Gram-negative versatile bacterium. From harmless inhabitant to pathogenic colonizer of humans and animals, this bacterium has been extensively studied [Kaper et al., 2004; Anjum et al., 2021; ECDC, EFSA Panel on Biological Hazards (BIOHAZ), and EMA Committee for Medicinal Products for Veterinary Use (CVMP), 2017]. Within pathogenic E. coli, different pathotypes have been identified resulting in a spectrum of clinical syndromes ranging from mild, self-limited gastroenteritis to urinary tract infections and meningitis (Kaper et al., 2004). Within intestinal pathogenic E. coli, enteropathogenic E. coli (EPEC) have been reported. As enteric bacterial pathogen, EPEC has been described as a relevant factor associated to diarrhea in Humans of pediatric age in developing countries (Gomes et al., 2016; Ochoa and Contreras, 2011). Recently, EPEC has re-emerged associated with sporadic human cases or outbreaks in both developed and developing Countries (lee et al., 2022; Platenkamp and Mellies, 2018). Regarding outbreaks, several food categories were pinpointed as vehicles of transmission identifying specific EPEC strains as foodborne pathogens. Rocket salad was identified as source of contamination in 42 confirmed cases of EPEC infection in 2016 in Finland with symptoms ranging from none to diarrhea, vomiting and fever (Kinnula et al., 2018). In South Korea, 13 children in an elementary school were infected after consumption of water contaminated foods such as seasoned cucumber and chives (Lim et al., 2020). Besides plant-based foods, also food of animal origin can be contaminated by EPEC. EPEC was detected in more than 50% of eviscerated chicken carcasses in Argentina as well as in frozen carcasses in Iraq (Alonso et al., 2011; Taha and Yassin, 2019). Moreover, samples of raw dairy and meat products of bovine origin were positive for EPEC in Iran (Abri et al., 2019).
At present, EPEC are identified based on their pattern of virulence genes. Similar to enterohemorrhagic Escherichia coli (EHEC), all EPEC carry the eae gene, coding for intimin and included in the Locus for enterocyte effacement (LEE) associated to the characteristic attaching and effacing lesion (Kaper et al., 2004). Differently from EHEC, all EPEC do not carry the stx1 and stx2 genes coding for Shiga toxins. EPEC can be additionally divided in two groups namely “typical” and “atypical” EPEC based on the presence or absence of the bfp gene, respectively. The bfp gene is essential for biosynthesis of bundle-forming pili and it is located in the EPEC adhesion factor (EAF) plasmid (Kaper et al., 2004). Typical EPEC are considered diarrheagenic strains especially in children, whereas more controversial is the pathogenicity of atypical (a)-EPEC which have been isolated from both diseased and healthy Humans (Slinger et al., 2017; Ochoa and Contreras, 2011).
Along with intestinal pathogenic E. coli, pathotypes of extraintestinal pathogenic E. coli (ExPEC) have been reported as relevant Human pathogens associated with a variety of outcomes from meningitis in neonates to bacteremia in older adults and urinary tract infections in young healthy women (Russo and Johnson, 2003). In foods, ExPEC were isolated commonly in poultry meat, as well as in pork and beef although with a less extent. Poultry, pork and, with a minor extent, beef, have been associated to ExPEC lineages which emerged in the 1990s and they are still accounting for a large fraction of ExPEC diseases in humans (Manges and Johnson, 2012; Meena et al., 2023). ExPEC ST131 was detected in poultry and UTI isolates in Canada as well as in pork and UTI in Danmark and Norway (Vincent et al., 2010; Trobos et al., 2009). Direct link of human UTI lineage ExPEC ST69 to food, primarily pork and chicken meat, was suggested by PFGE similarities and pathogenicity assessment of food isolates in in vivo UTI mouse model (Vincent et al., 2010; Jakobsen et al., 2012). PFGE similarities were encountered between clinical and cow samples of ExPEC ST69 in US (Ramchandani et al., 2005). Multidrug-resistant ExPEC ST10 isolates were retrieved from human clinical samples, retail chicken and pork meat (although with limited PFGE similarity) in Canada (Bergeron et al., 2012). Regarding the molecular bases of ExPEC pathotype, several virulence factors were identified: (1) adhesins (i.e., papACEFG—P fimbriae), (2) iron acquisition systems (i.e., iuc—aerobactin), (3) protectins (i.e., *kpsM II—*kpsM II group 2 capsule), (4) invasins (i.e., ibeABC—cell invasion into the host tissue) and (5) toxins [i.e., hylA—α-haemolysin, cytotoxic necrotizing factor (cnf) and cytolethal distending toxin (cdt); Sora et al., 2021; Sarowska et al., 2019; Dale and Woodford, 2015; Onlen Guneri et al., 2022].
Although several virulence factors have been described in both aEPEC and ExPEC, the prediction of the final phenotypic outcome is challenging due to the high complexity of the virulome and the potential differential expression of its genes (Peirano et al., 2013). In this context, a particular relevance is covered by in vivo infection models essential to evaluate the pathogenic outcome of strains with different virulence patterns. Along with mouse and chicken animal models, other non-vertebrates animal models have been recently arising such as the larvae of the greater wax moth Galleria mellonella (Serrano et al., 2023; Rehman et al., 2017; Green et al., 2015; Antão et al., 2008; Ménard et al., 2021). In particular, this invertebrate has increasingly been used as an alternative model host in microbial pathogenesis studies. Adhering to the 3R principle of replacement, reduction and refinement, Galleria mellonella, as an invertebrate infection model, is not subject to Directive 2010/63/EU on the protection of animals used for scientific purposes. With its innate immune system very similar to mammal’s one, larvae of G. mellonella provides unprecedent opportunity for a first evaluation (or confirmation) of pathogenicity of bacterial strains. The translatability of results was documented by Velikova and colleagues, who observed that the pathogenicity in G. mellonella of different serotypes and mutants of Streptococcus suis was in agreement with pathogenicity observed in piglets (Sus scrofa; Velikova et al., 2016). Additional studies suggested the utility of this in vivo model to evaluate the pathogenic potential specifically of ExPEC and EPEC (Ménard et al., 2021; Williamson et al., 2014; Leuko and Raivio, 2012; Pasquali et al., 2025).
In a previous study, genomes of 33 E. coli isolates collected from Italian artisanal food productions were sequenced and their virulome and resistome characterized. Virulence gene patterns predicted putative pathogenicity for three genomes (Crippa et al., 2024). In the present study, the pathogenicity potential of previously sequenced E. coli was further investigated by: (1) phylogenomic comparison with 202 public genomes of human, animal and environmental origin; (2) pathogenicity assessment of strains with virulence patterns predicting specific E. coli pathotypes by using larvae of Galleria mellonella as in vivo infection model.
Materials and methods
2
Rationale for isolate selection
2.1
In a prior investigation, a total of 1,170 samples were collected between January 2020 and May 2021 from raw materials, semi-finished and finished products, as well as environmental surfaces, across six production cycles at two artisanal facilities producing soft cheese and organic salami in Italy (Pasquali et al., 2022; Pasquali et al., 2023). Biotyping analysis identified Escherichia coli in three soft cheese samples (one semi-finished and two finished products) and in 30 samples from the salami production line. The latter included raw materials, semi-finished products aged for 18 weeks, and surfaces and equipment such as tables and fillers located in the processing environment (Crippa et al., 2024).
Whole genome sequencing
2.2
In order to investigate the virulome of 33 E. coli isolates, the DNA was previously extracted using the MagAttract HMW DNA Kit (Qiagen, Milan, Italy) and sequenced on a Illumina NovaSeq platform (Illumina, Milan, Italy). Reads were assembled de novo using Unicycler v0.5.0 (Wick et al., 2017), and the assembled contigs were screened with ABRicate v1.0.1 (Seemann, 2020) using ResFinder (Bortolaia et al., 2020) and ecoli_VFdatabases (Crippa et al., 2024). In the present study, for comparison purposes to published papers, sequence types (STs) were assigned using mlst v2.23.0 (Seemann, 2022), based on the PubMLST database (Jolley and Maiden, 2010), whereas O: H serotypes and phylogroups were predicted in silico using the EnteroBase typing tools (Zhou et al., 2020).
SNP-based phylogenetic analysis
2.3
In order to infer phylogenetic relationships among E. coli strains isolated from artisanal food of animal origin and E. coli strains from other sources (human, environment, animal) SNP calling was performed incorporating both the 33 Italian E. coli strains and 202 publicly available E. coli genomes sourced from Enterobase (Dyer et al., 2025). Public genomes were selected based on Country (Italy), Collection year: 2020–2023, and their metadata including serotypes, STs and lineages were retrieved from the Database (Supplementary Table S1). SNP identification and tree construction were, respectively, carried out with Snippy v.4.6.0 (Seemann, 2015) and PhyML v.3.1 (Guindon et al., 2010), using the reference genome Escherichia coli str. K-12 substr. MG1655 (NCBI RefSeq assembly no. GCF_000005845.2). The latter was implemented to build a maximum likelihood phylogenetic tree based on an alignment of core SNPs produced with snippy-core. The tree was visualized and annotated with metadata using iTOL v6 (Letunic and Bork, 2024). Pairwise SNP distances were calculated with snp-dists v0.6.3 (Taouk et al., 2025).
Virulome
2.4
To assess genetic determinants of virulence, all 235 assembled contigs were screened with ABRicate v1.0.1 (Seemann, 2020) using ecoli_vf database (Chen et al., 2016), setting a minimum coverage threshold of 90% and identity 60%. The presence/absence heatmap of virulence-associated genes identified in newly sequenced E. coli was visualized using the R package pheatmap v1.0.13. Among all genes detected by the database, only those typically described for ExPEC (Sarowska et al., 2019; Dale and Woodford, 2015) and EPEC (Bugarel et al., 2011; Perna et al., 2001; Garmendia et al., 2005; Karmali et al., 2003; Konczy et al., 2008) pathotypes were included in the heatmap. Moreover, samples were annotated by source type and sample origin.
To explore source-related differences in virulence genes among Escherichia coli isolates, statistical analysis was performed using R v4.5.0. Specifically, the prevalences of genes malX and cnf1, specifically found in genomes of putative EXPEC strains showing higher pathogenicity in G. mellonella, were compared between isolates of human and other (food, environment and animal) origin. The expected counts were calculated using the default method implemented in R’s chisq.test () function, based on marginal totals under the assumption of independence. Pearson’s chi-square test (χ2) was applied for malX as all expected cell counts were ≥ 5, whereas Fisher’s exact test was used for cnf1 due to the presence of expected cell counts < 5. Odds ratios (ORs) with 95% confidence intervals (CIs) were calculated to quantify the strength of associations. Statistical significance was set at α = 0.05, without corrections for multiple testing.
Pathogenicity assessment
2.5
Pathogenicity was evaluated using Galleria mellonella larvae (Terraqua, Torino, Italy). Final-instar larvae (200–250 mg) were infected following the protocol by Gallorini et al. (2024). Bacterial cultures were prepared from Mueller Hinton Agar plates, then transferred to Mueller Hinton II broth and incubated at 37 °C for 16 h. Cells were centrifuged, washed in PBS, and resuspended. Optical density at 600 nm was used to standardize cell concentrations. Preliminary studies were performed with different concentrations ranging from 10^3^ to 10^8^ CFU per 10 μL. Bacterial suspensions of approx. 10^4^ CFU/10 μL were selected since they corresponded to the mortality of 50% of larvae after 1 day post inoculum (L50; data not shown) and were injected into the third left pro-leg of each larva. Serial dilutions were plated to confirm inoculum size, estimated at 5.71–5.95 log₁₀ CFU/mL. Eight strains were tested in triplicate with 10 larvae per replicate. Controls included PBS-injected larvae, non-injected larvae as well as larvae injected with a predicted hypovirulent strain (1SBR281). In total, 300 larvae were used. Infected larvae were incubated at 35 °C in darkness and monitored daily over 5 days for survival. Data were analyzed via the Kaplan–Meier estimator which was employed to determine the survival function, estimating the cumulative probability of G. mellonella larval survival over the five-day observation period. This non-parametric method correctly accounts for censored data (larvae that survived the full duration) by calculating the probability of survival at each observed death time based on the number of individuals at risk (Kaplan and Meier, 1958). The results are presented visually as Kaplan–Meier survival curves, and statistical differences among groups were assessed by the log-rank test using the survival package in R v4.3.2. A p-value < 0.05 was considered statistically significant. Moreover, pairwise comparisons between survival curves were performed using the log-rank test implemented in the survminer R package, with p-values adjusted for multiple testing using the Benjamini–Hochberg false discovery rate (FDR) correction.
Results
3
Escherichia coli taxonomic assignment and de novo assembly
3.1
A total of 1,170 food and environmental samples were previously collected within 6 consecutive batches in the two artisanal productions of soft cheese and salami (Pasquali et al., 2022; Pasquali et al., 2023). Based on bio-typing results, E. coli was detected in 33 samples: 3 from soft cheese semifinished and finished food products and 30 from salami production (raw materials, semi-finished and finished food products, environmental samples). Following whole genome sequencing, the genomes of those isolates were confirmed as belonging to Escherichia coli with percentages of Average Nucleotide Identity (ANI) between 98.73 and 99.94% (Table 1).
Draft genomes showed good quality statistics: low number of contigs (52–172) and high N50 (91064–553,490). Genome length (4.7–5.2 Mb) and GC contents (50.4–50.8%) were in the typical range for Escherichia coli (Crippa et al., 2024; Criscuolo, 2018).
Phylogenetic analyses
3.2
In order to decipher the genetic relationship of the newly sequenced E. coli with isolates previously collected in Italy in diseased humans, food, animals and the environment, a maximum likelihood (ML) phylogenetic tree was inferred including 202 public genomes along with the 33 E. coli genomes of artisanal food origin (Figure 1). Among the 33 E. coli genomes high genetic distances were observed between the genomes collected from the processing environment and genomes collected from food as well as within isolates collected from food (Table 1; Figure 1; Supplementary Figure S1). In contrast, in the salami production, 3 of the five genomes of isolates collected from raw materials (pig meat mixture), namely 2 MB1, 3 MB1 and 4 MB1, showed close genetic proximity to genomes of isolates collected from food 2SBR4, 3SBD4, 2SBR282 with 26, 23 and 33 SNPs difference, respectively. This observation suggests the absence of persistent clones as well as raw material and not the processing environment as potential source of contamination of salami. Interestingly, when considering also public genomes, genome 3SBR3 (E. coli isolated from semifinished salami) was gathered in cluster ECO1 including also two public genomes of E. coli isolated from wild boar in Italy in 2021 and 2022 (assembly barcode ESC_NC7896AA_AS and ESC_NC7887AA_AS, 648 and 655 SNPs difference respectively; Figure 1; Supplementary Figure S1). This observation reinforces the suggestion of pigs and wild boars as reservoirs of E. coli. Moreover, genomes 6 MB5 (raw material) and 5STM5 (salami processing environment) fell within cluster ECO2 gathering 9 genomes of clinical relevance (7 isolates collected from urine, 1 from blood and 1 from wound) and 7 genomes of wild boar. Although the relevant SNP difference among genomes of this cluster (ranging from 26,260 to 37,851 SNPs difference; Figure 1; Supplementary Figure S1), the inclusion of 6 MB5 and 5STM5 in this cluster reinforces the genetic similarity of these genomes to genomes isolated from extraintestinal human sites suggesting pigs and wild boars as potential reservoirs of pathogenic E. coli (Figure 1).
Maximum likelihood phylogenetic tree inferred from core gene alignments of 33 Escherichia coli (in bold) isolated from two Italian meat and dairy artisanal food productions and 202 public genomes of E. coli isolated in Italy from 2020 to 2023 from humans, food, animals, and the environment. The tree was rooted with reference Escherichia coli str. K-12 substr. MG1655 (NCBI RefSeq assembly no. GCF_000005845.2). Clusters ECO1 and ECO2 are highlighted in yellow and green branches, respectively.
Virulome
3.3
The investigation of virulence associated genes in newly sequenced E. coli genomes, revealed different patterns with number of virulence genes ranging from 143 to 254. Of interest, E. coli genomes 5STM5 and 6 MB5, which were found closely genetically related to public genomes of clinical relevance, were carriers of virulence genes belonging to ExPEC and/or aEPEC pathotypes.
In particular*, E. coli* strains 5STM5 (−: H49, ST4102), along with 4STM3 (O108: H21, ST337), and 6STM2 (O45: H2, ST301), were isolated from the processing environment of the artisanal salami (surface of the table in the stuffing room; Table 1; Figure 1). All three strains harbored the eae and tir genes within the locus of enterocyte effacement (LEE) and they were negative for the bftp as well as for the stx1 and stx2 genes thus belonging to atypical enteropathogenic E. coli following previous categorization (Figure 2; Levine et al., 1985). In particular they belong to phylogroups B1, F and A and to serogroups O108: H21, −: H49, and O45: H2, respectively.
Heatmap of virulence associated genes of the 33 E. coli isolated from artisanal food productions. Only virulence genes already reported as associated to the EPEC (Bugarel et al., 2011; Perna et al., 2001; Garmendia et al., 2005; Karmali et al., 2003; Konczy et al., 2008) and ExPEC pathotypes (Sarowska et al., 2019; Dale and Woodford, 2015) are displayed. Colors black and white represent percentage of proportion of gene covered with reference virulence gene sequences.
Moreover, although negative for the lifA gene, all three genomes carried virulence genes belonging to genetic islands OI-71 (nleH, nleA, nleF) and OI-122 (nleB, nleE, espL). Interestingly, all three genomes carried additionally the traT gene. This gene codes for a lipoprotein located on the outer membrane of E.coli and involved in the adhesion to host cells.
Another interesting observation is that among genes described as virulence factor of EPEC, the espC gene was detected in genome 5STM5 (Figure 2; Supplementary Table S2). This genome was the only one carrying this gene among the 235 genomes analyzed, confirming the low frequency already reported previously (Afset et al., 2006; Hernandes et al., 2020; Ocampo et al., 2021).
Of interest, genome 5STM5 carries the eae gene along with additional genes generally listed as ExPEC virulence markers namely chuA, csgA, ecpA, fimH, ibeA, iss2, iucA iutA, ompA, ompT and traT (Sarowska et al., 2019; Dale and Woodford, 2015; Figure 2). The co-occurrence of the eae gene along with these genes suggest this strain as hybrid aEPEC/ExPEC. This unusual and singular genetic combination might lead to higher virulence due to the presence of virulence factors from two different pathotypes.
Along with aEPEC strains, additional attention was pointed toward 6 MB5 as well as other three E. coli strains: one isolated from raw materials (6 MB5 O2: H6, ST141), two from semifinished products of the ripening room (2SBR183 O5: H4, ST93; 3SBR181 D12: H4, ST57) of the salami production, and one from the cheese final product (3CP1522 O45: H8, ST 297). The genomes of these strains carried from 13 to 26 virulence genes previously reported in extraintestinal pathogenic E. coli such as, among others, hlyA, cnf1, iroN, iss2, iucA, iutA, KpsM, papC and malX (Sora et al., 2021; Sarowska et al., 2019; Dale and Woodford, 2015; Figure 2). The highest number of ExPEC associated virulence genes was found in E. coli strain 6 MB5.
In vivo pathogenicity assessment in galleria mellonella larvae
3.4
In order to assess the pathogenicity of presumptive aEPEC and ExPEC, Galleria mellonella larvae were tested as in vivo infection model. In particular, all eae and tir positive strains (4STM3, 5STM5 and 6STM2) were tested as presumptive aEPEC. Additionally, strains 6 MB5, 2SBR183, 3SBR181, 3CP1522 were tested by the in vivo approach. Although, the virulence gene pattern definition of ExPEC pathotype is still unclear, these four strains were selected for one or both of the following reasons: (1) they harbored virulence genes already described in ExPEC pathotype, (2) they showed genetic similarities with public human genomes of clinical relevance. In order to evaluate the impact on Galleria mellonella mortality of E. coli strains with different virulence patterns, and to assess the immune stimulation caused by a high bacterial load of a hypovirulent strain, the strain 1SBR281 with the lowest number of virulence genes associated to ExPEC and EPEC pathotype, was also included.
After 5 days post infection, with an inoculum of 10^4^ CFU/10 μL, the highest percentage of larvae mortality among aEPEC presumptive strains, was observed in relation to challenge test with strain 5STM5. In particular, 5STM5, 4STM3 and 6STM2 were associated to survival of infected Galleria mellonella of 50.0%, 66.6, and 70%, respectively (Figure 3). Log-rank test analysis confirmed a statistically significant difference in survival among EPEC strains (Chi-square = 22.9, df = 5, p 3 × 10–4), showing from pairwise comparison 5STM5 mortality rate as significantly higher compared to 1SBR281 (p < 0.05; Supplementary Table S3). Actually, the predicted hypovirulent strain 1SBR281 showed indistinguishable mortality curve in comparison to not-infected larvae (CONTROL; Figure 3; Supplementary Table S3).
Kaplan–Meier plot showing percentage survival of Galleria mellonella larvae after inoculation with bacterial suspensions of E. coli strains representative of identified virulence patterns associated to aEPEC pathotype. Noninjected larvae (Control) and larvae injected with sterile PBS (PBS) are included. For each treatment, n = 30 (pooled from triplicate experiment).
Among the presumptive ExPEC strains, the survival rates after 5 days post infection were 13,3, 30, 60 and 70% for 3CP1522, 6 MB5, 2SBR183 and 3SBR181, respectively (Figure 4), with significat difference highlighted by long-rank test (Chi-square = 92.5, df = 5, p < 2 × 10–16). The high mortality rate of larvae infected by 3CP1522 and 6 MB5 was statistically significant different in comparison to the mild mortality associated to 2SBR183 and 3SBR181 (p < 0.05; Supplementary Table S3). These results suggest the complex pathway of genotype to phenotype correspondance and it reinforces the relevance of in vivo models to assess aEPEC and ExPEC presumptive pathogenicity which seems not to be fully predictable by the genotype alone.
Kaplan–Meier plot showing percentage survival of Galleria mellonella larvae after inoculation with bacterial suspensions of E. coli strains representative of identified virulence patterns associated to putative ExPEC pathotype. Noninjected larvae (CONTROL) and larvae injected with sterile PBS (PBS) are included. For each treatment, n = 30 (pooled from triplicate experiment).
Discussion
4
All 33 E. coli isolates collected from food and food processing environments in two artisanal productions of food of animal origins were sequenced The limited number of genomes prevents any consideration on seasonal variations or environmental effects, However, these data suggest the usefulness of WGS for traceability of contamination within the food processing environment and identification of raw materials as potential sources of contamination among the salami production. Among this food production, three E. coli strains showed virulence patterns specific of atypical Enteropathogenic E. coli pathotype being bfp-, stx1-, stx2-, eae+, tir+. Specifically, the eae gene of isolates 4STM3 (O108: H21, ST337), 5STM5 (−: H49, ST4102) and 6STM2 (O45: H2, ST301), belonged to subtypes Int-θ, Int-ι and Int-ε, respectively. Those three subtypes are considered to have a lower occurrence in comparison to more frequently identified subtypes like Int-β (Ramachandran et al., 2003). The identification of putative aEPEC in artisanal salami production is a public health concern not only for food consumers but also for workers involved in the preparation of salami in the stuffing room where these pathogens were isolated. Moreover, all three genomes carried virulence genes belonging to genetic islands OI-71 (nleH, nleA, nleF) and OI-122 (nleB, nleE, espL), already associated with clinical outcomes of infection (Karmali et al., 2003; Bugarel et al., 2011; Konczy et al., 2008). Interestingly, the co-occurrence of traT and eae gene has been recently described in E. coli exclusively of pig origin, confirming pig as reservoir of potential aEPEC strains of specific lineage (eae+, traT+; James et al., 2025). More investigations into a higher number of strains should be performed to confirm this hypothesis.
Another interesting observation is on the espC gene detected in genome 5STM5, which encodes for a protein secreted independently from the type III secretion system (Mellies et al., 2001). The gene is located on a genomic island, and it is not under the regulation of the type III secretion system as other esp genes (Mellies et al., 2001). Protein EspC belongs to class-1 of a superfamily of virulence factors named Serine Protease Autotransporters of Enterobacteriaceae (SPATE). It acts as autotransporter and it has enterotoxin activity playing a crucial role in the cytopathic effects on intestinal epithelium during EPEC infection (Navarro-Garcia, 2023). The frequency of EspC has been described as generally low. In a previous study in Norway 8 out of 57 aEPEC strains carried the gene (Afset et al., 2006). This frequency is consistent with a more recent study which reported a frequency of 8% in aEPEC genomes from all over the World (US, Africa, Europe, South Asia; Hernandes et al., 2020). Frequencies lower than 10% were reported also in another study from Peru (Ocampo et al., 2021). Interestingly in Brazil the reported occurrence has been higher (from 28 to 49% in aEPEC and tEPEC; Abreu et al., 2013; Hernandes et al., 2020). The present study confirmed the low frequency of this gene in presumptive aEPEC as well as in other E. coli, with only one positive genome among the 235 Italian E. coli genomes analyzed. In genome 5STM5 the espC gene is located in a unique DNA region of the chromosome downstream of genes glaH, ygaF (renamed lhgO) and operon gabDTP. This localization has been previously reported in a espC+ pathogenic EPEC strain E2348/69 (O127: H6; Mellies et al., 2001). Further studies should be performed to confirm the selective advantage of strains carrying the espC gene in the survival and pathogenicity of E. coli in humans.
Moreover, the co-occurrence of the eae gene along with extraintestinal pathogenic E. coli (ExPEC) virulence markers chuA, csgA, ecpA, fimH, ibeA, iss2, iucA iutA, ompA, ompT and traT (Sarowska et al., 2019; Dale and Woodford, 2015) suggest the 5STM5 strain as hybrid aEPEC/ExPEC. This unusual and singular genetic combination might lead to higher virulence due to the presence of virulence factors from two different pathotypes. Hybrid strains have been previously reported. Among others, the notable strain E. coli O104: H4, associated to the multi-Country outbreak of 2011, was a hybrid pathogen that carried virulence genes found in both typical enteroaggregative E. coli (EAEC) strains (aggA, aggR, set1, pic, and aap) and Shiga-toxin producing E. coli (STEC; stx 2; Croxen et al., 2013). More specifically, a hybrid aEPEC/ExPEC isolate was collected in Brazil from feces of a patient with acute diarrhea and firstly assigned as EPEC. After sequencing, the genome of the isolate revealed the presence of LEE genes along with 16 virulence genes more frequently identified in ExPEC strains (Munhoz et al., 2021). Another hybrid aEPEC/UPEC was collected from patient with urinary tract infection in Italy and firstly assigned as UPEC. After sequencing, authors noticed the presence of the LEE, which genes espB and eae were confirmed as expressed (Valiatti et al., 2020).
Concerning the in vivo pathogenicity assessment in G. mellonella larvae, results of survival rates reinforced the genomic predictions of higher virulence of aEPEC strain 5STM5 and pointed out to an absent or mild pathogenic potential of aEPEC strains 4STM3 and 6STM2. The mild pathogenicity potential of strain 4STM3, is reinforced by its phylogenetic proximity to the human strain with assembly barcode ESC_PC5093AA_AS isolated previously from a healthy donor (Figure 1; Di Pierro et al., 2024). Mild to non-pathogenic aEPEC have been already described worldwide (Hernandes et al., 2009; Nguyen et al., 2006). On the other hand, the mild pathogenicity encountered for 6STM2 is surprising since this strain belongs to ST301 clonal complex and to serogroup 045: H2 already described as gathering shiga toxin producing Escherichia coli (Zhang et al., 2020).
Along with aEPEC putative strains, four strains showed interesting virulence patterns being positive for several genes already described in ExPEC pathotypes. Although virulence genes of ExPEC have been described, there is not at present an agreed identification of key virulence markers essential for the observation of this pathotype, making difficult the prediction of the pathogenic potential of these strains solely based on their virulome.
Among the presumptive ExPEC strains, the two with the highest mortality rate in G. mellonella larvae (3CP1522 and 6 MB5) carried the malX gene which was absent in the genomes of the other strains tested. This gene is a virulence marker of the pathogenicity Island of strain CFT073 and has been described as an epidemiological predictor of ExPEC specifically of urinary and pulmonary source (Johnson and Russo, 2018; Johnson et al., 2002). Additionally, it was found in an epidemic clone of multiresistant ESBL ExPEC in Denmark (Olesen et al., 2013). In the present study the malX gene showed a significantly higher occurrence in humans vs. other (food, environmental, animal) sources (65% vs. 17%, p-value = 4.958e-07), substantiating its role in ExPEC pathotype. The 6 MB5 genome carried additionally the cnf gene coding for the cytotoxic necrotizing factor (CNF), which has been traditionally considered as a ExPEC virulence gene marker thanks also to confirmatory evidence in animal model infection studies (Khan et al., 2002; Rippere-Lampe et al., 2001). As for malX, in the present study also the occurrence of cnf1 gene was higher in isolates of clinical origin vs other origins (30% vs 3%) although this difference was not statistically significant (p-value = 0.2466). Surprisingly, although representing the genome with the highest number of ExPEC virulence strains, 6 MB5 strain was not the strain with the highest pathogenicity in G. mellonella reinforcing the complexity of ExPEC phenotype prediction based on the virulome. Whole genome sequencing is effective in detecting virulence genes but is not informative on their expression. A phenotypic virulence might be affected by gene regulation, non-functional genes or complex interactions among genes and proteins. Genotypic to phenotypic discordances have been fully described for antimicrobial resistance (Yee et al., 2021). These discrepancies further reinforce the relevance of an in vivo infection model to phenotypically explore the pathogenicity potential of strains with specific virulence patterns. This study has limitations. Along with the limited number of strains tested, results on an in-vivo infection model should be considered with caution when translability to humans is considered. Moreover, concerning the highlighted espC, malX and cnf1 genes, a specific quantitative risk assessment is required to evaluate the real impact on consumer health risk. Further studies are needed to confirm the relevance of these genes on aEPEC and ExPEC pathotypes.
Conclusion
5
In conclusion, the combined genomic and in vivo approach used in the present study was useful to evaluate the potential pathogenicity of E. coli strains isolated from artisanal salami and soft cheese helping in pinpointing to specific aEPEC, ExPEC and hybrid aEPEC/ExPEC strains which showed higher mortality in G. mellonella larvae and were characterized by additional virulence genes such as eae and espC in putative aEPEC, and malX in putative ExPEC. The potential concern for public health associated to these strains, and in particular the direct role of each of these genes alone and in combination with others, is worthy of future investigations which could help to assess the risk for consumers after the consumption of contaminated artisanal food.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Abreu A. G. Bueris V. Porangaba T. M. Sircili M. P. Navarro-Garcia F. Elias W. P. (2013). Autotransporter protein-encoding genes of diarrheagenic Escherichia coli are found in both typical and atypical enteropathogenic E. coli strains. Appl. Eviron. Microbiol. 79, 411–414. doi: 10.1128/AEM.02635-12, 23104414 PMC 3536084 · doi ↗ · pubmed ↗
- 2Abri R. Javadi A. Asghari R. Razavilar V. Salehi T. Z. Safaeeyan F. . (2019). Surveillance for enterotoxigenic and enteropathogenic Escherichia coli isolates from animal source foods in Northwest Iran. Indian J. Med. Res. 150, 87–91. doi: 10.4103/ijmr.IJMR_2019_17, 31571634 PMC 6798612 · doi ↗ · pubmed ↗
- 3Afset J. E. Bruant G. Brousseau R. Harel J. Anderssen E. Bevanger L. . (2006). Identification of virulence genes linked with diarrhea due to atypical enteropathogenic Escherichia coli by DNA microarray analysis and PCR. J. Clin. Microbiol. 44, 3703–3711. doi: 10.1128/JCM.00429-06, 17021100 PMC 1594803 · doi ↗ · pubmed ↗
- 4Almli V. L. Verbeke W. Vanhonacker F. Næs T. Hersleth M. (2011). General image and attribute perceptions of traditional food in six European countries. Food Qual. Prefer. 22, 129–138. doi: 10.1016/j.foodqual.2010.08.008 · doi ↗
- 5Alonso M. Z. Padola N. L. Parma A. E. Lucchesi P. M. (2011). Enteropathogenic Escherichia coli contamination at different stages of the chicken slaughtering process. Poult. Sci. 90, 2638–2641. doi: 10.3382/ps.2011-01621, 22010252 · doi ↗ · pubmed ↗
- 6Anjum M. F. Schmitt H. Börjesson S. Berendonk T. U.WAWES network (2021). The potential of using E. coli as an indicator for the surveillance of antimicrobial resistance (AMR) in the environment. Curr. Opin. Microbiol. 64, 152–158. doi: 10.1016/j.mib.2021.09.011, 34739920 · doi ↗ · pubmed ↗
- 7Antão E. M. Glodde S. Li G. Sharifi R. Homeier T. Laturnus C. . (2008). The chicken as a natural model for extraintestinal infections caused by avian pathogenic Escherichia coli (APEC). Microb. Pathog. 45, 361–369. doi: 10.1016/j.micpath.2008.08.005, 18848980 · doi ↗ · pubmed ↗
- 8Assolatte. (2025). Rapporto ASSOLATTE 2025. Available online at: https://www.assolatte.it/it/home/news_detail/attualita/1751433709593 [Accessed on the 24th October 2025]