Genomic characterization and prognostic significance of copy number alterations in Tunisian patients with acute lymphoblastic leukemia
Ameni Bedoui, Wajdi Ayadi, Nour Louati, Imen Frikha, Yosra Fakhfakh, Fahmi Smaoui, Ali Gargouri, Ikram Ben Amor, Moez Elloumi, Moez Mdhaffar, Raja Mokdad Gargouri

TL;DR
This study identifies key genetic changes in Tunisian ALL patients and shows how these changes affect treatment response and survival.
Contribution
The study provides new insights into the role of IKZF1 deletions in ALL prognosis and treatment response in a Tunisian population.
Findings
IKZF1 deletions were significantly linked to poor glucocorticoid response and higher MRD rates in B-ALL patients.
Patients with IKZF1 deletions had significantly lower survival rates in both univariate and multivariate analyses.
IKZF1 deletion status could improve risk stratification models for ALL patients.
Abstract
Acute lymphoblastic leukemia (ALL) is a heterogeneous malignancy characterized by various genomic alterations playing a crucial role in disease classification, prognosis, and response to treatment. However, molecular diagnosis and effective management of this hematological malignancy remain a major challenge, particularly in developing countries, including Tunisia. In this study, we aimed to conduct a detailed analysis of copy number alterations (CNAs) associated with ALL in a cohort of 60 primary samples from Tunisian patients. Using multiplex ligation-dependent probe amplification (MLPA), major genetic lesions, including IKZF1, CDKN2A/2B, PAX5, ETV6, BTG1, and genes located in the PAR1 region, were analyzed and their associations with clinical and laboratory features, as well as survival outcomes, were also evaluated. Our analysis revealed that 70% of patients had deletions and/or…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
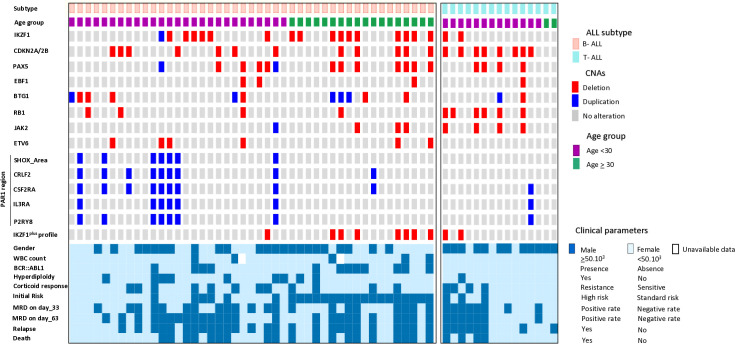 Fig 1
Fig 1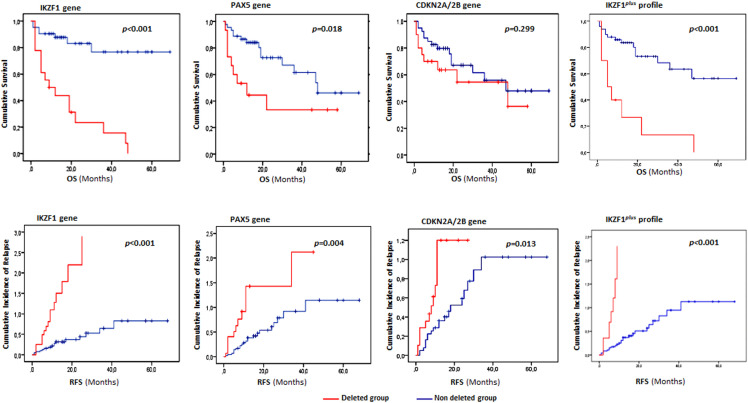 Fig 2
Fig 2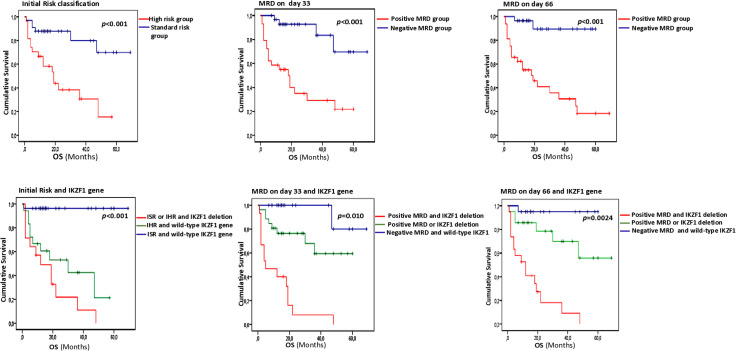 Fig 3
Fig 3Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAcute Lymphoblastic Leukemia research · Genomics and Rare Diseases · Acute Myeloid Leukemia Research
Introduction
Acute lymphoblastic leukemia (ALL) is a malignancy of B- or T-lineage lymphoblast’s, characterized by the abnormal clonal expansion of immature progenitor cells in the bone marrow (BM). These leukemic cells can infiltrate the peripheral blood (PB) and disseminate to other organs, including the central nervous system, lymph nodes, liver, and spleen [1–3]. ALL is a common childhood malignancy, accounting for up to 25% of childhood cancers. It can also occur in adulthood, with variable clinical, biological, and molecular features across age groups [4,5]. The incidence of ALL varies considerably among countries [6]. Globally, the estimated age-standardized incidence rate increased from 1.23 per 100,000 in 1990 to 1.96 in 2019, with a marked rise in cases among older individuals, particularly in populations with a higher socio-demographic index [7]. According to the latest Tunisian Cancer Registry, ALL accounts for 45.1% of all hematological malignancies, with a standardized incidence rate of 2.25. B-ALL is the most common subtype, representing 71% of cases, of which 78% occur in pediatric/young adult patients. In contrast, T-ALL accounts for 29% of cases, a proportion that appears higher than that reported in European populations [8]. Over the past few decades, the management of ALL has evolved considerably, leading to notable improvements in survival and cure rates, particularly among pediatric patients. This progress is mainly attributable to major advances in molecular diagnostics, risk stratification, and the approval of targeted therapies [9]. Despite these advancements, relapse, which affects approximately 15–20% of patients, remains a major clinical challenge, particularly in low- and middle-income countries, where access to advanced diagnostics and therapies is often limited [10].
Although the exact etiology of ALL is still not fully understood, ongoing research continues to elucidate the genetic abnormalities that impair hematopoiesis and promote leukemogenesis. Interestingly, several genetic alterations have emerged as valuable prognostic and therapeutic markers, playing a critical role in risk stratification and guiding appropriate treatment strategies. Adverse prognostic indicators include fusion genes such as BCR::ABL1 and MLL::AFF1, which are associated with a high risk of relapse and poorer outcomes [11–13]. In contrast, favorable prognostic markers such as ETV6::RUNX1 and high hyperdiploïdy are correlated with better treatment responses and long-term survival [14,15].
More recently, the use of next-generation sequencing (NGS) and multiplex ligation-dependent probe amplification (MLPA) has significantly advanced our understanding of molecular pathogenesis, defining a broad spectrum of genetic abnormalities, including CNAs, particularly micro-deletions in genes involved in B-lymphocyte development [16–18]. One of the most clinically relevant genes is IKZF1 (Ikaros family zinc finger 1), which encodes a transcription factor essential for the development and function of lymphocytes, especially B-cell precursors [19,20]. IKZF1 plays a pivotal role in regulating the immune system by controlling the differentiation and activity of various immune cell types, including B- and T-lymphocytes [19–21].
Besides IKZF1, particular attention has been given to CNAs affecting other genes, including ETV6, PAX5, and EBF1, essential for B-cell differentiation, as well as CDKN2A/2B, BTG1, and RB1, involved in cell cycle regulation and tumor suppression [4,18,22,23]. Furthermore, CNAs disrupting cytokine receptor genes such as CRLF2, IL3RA and CSF2RA located in the pseudoautosomal region 1 (PAR1) region have also been identified to be associated with aberrant activation of cytokine-mediated signaling pathways, which play a critical role in leukemogenesis [24,25]. In fact, CRLF2 gene overexpression often results from PAR1 rearrangements, such as IGH::CRLF2 translocations or P2RY8::CRLF2 fusions, leading to constitutive activation of the JAK-STAT signaling pathway, which promotes leukemic cell proliferation and survival [24–26]. Although most of these gene alterations demonstrate prognostic relevance in ALL patients, deletions in IKZF1 have emerged as particularly clinically significant [27,28]. This has led to the integration of IKZF1 status into modern risk stratification schemes and highlighted its utility in identifying patients who might benefit from intensified therapeutic approaches [29,30]. However, the optimal treatment strategy for ALL patients with IKZF1 deletions remains controversial, with ongoing debate regarding the use of IKZF1 deletion status as a basis for high-risk stratification.
To support these advances, the present study aimed to provide a detailed analysis of the broader spectrum of genetic alterations associated with ALL, as well as clinical relevance and prognostic significance in Tunisian patients. Such valuable information could support the continued monitoring of progress in risk stratification criteria, with a view to more tailored and individualized therapeutic decision-making.
Materials and methods
Study cohort
Bone marrow and/or peripheral blood samples containing more than 60% blasts were retrospectively collected from 60 patients newly diagnosed with ALL at the Hemobiology Laboratory of the Regional Blood Transfusion Center in Sfax, Tunisia, between January 2020 and March 2024. Samples were obtained as part of routine clinical procedures for biological diagnosis, and only the residual material not required for diagnostic purposes was used in the present study. Patients’ ages ranged from 1 to 72 years; 40 were under 30 (pediatric/young adult form) and 20 were over 30 (adult form). The diagnosis of precursor cell origin was based on the conventional FAB (French–American–British) classification and immunophenotypic criteria; 45 patients (75%) were diagnosed with B-cell precursor ALL, and 15 patients (25%) with T-cell precursor ALL. The main characteristics and genetic features of the study cohort are shown in Table 1. Ethical approval was granted by the local Ethics Committee of the Faculty of Medicine in Sfax (approval no. 12/25, dated February 14, 2025). The samples were fully anonymized before being accessed on February 15, 2025, for research purposes. The Ethics Committee waived the requirement for informed consent because no identifiable information about participants was available after data collection.
Table 1: Clinical and genetic features of the ALL studied cases (n = 60).
As internal controls for methodological standardization of the MLPA assay, 15 samples from healthy individuals were collected between December 2020 and March 2023, following written informed consent. The control samples were obtained from volunteer blood donors with no prior clinical history or relevant medical conditions, referred to the Hemobiology Laboratory of the Regional Blood Transfusion Center in Sfax, Tunisia.
Therapy groups and risk stratification
Patients were treated at the Clinical Hematology Department of the Hedi Chaker University Hospital in Sfax, Tunisia, according to the EORTC-CLG 58951 protocol (European Organization for Research and Treatment of Cancer), developed for pediatric and young adult patients, or the GRALL-2014 protocol (Adult Acute Lymphoblastic Leukemia Research Group), designed for the management of adult patients over 30 years old. Philadelphia chromosome-positive (Ph+) patients were treated according to the GRAAPH-2005 protocol, which combines a tyrosine kinase inhibitor (TKI) with intensive chemotherapy. Minimal Residual Disease (MRD) analysis was performed by flow cytometry (FCM) on a Becton Dickinson FACS Canto II® cytometer. Follow-up bone marrow samples were collected on days 33 and 63, with a minimum of 100,000 events acquired per sample.
Based on the following criteria: normal bone marrow morphology (<5% blasts and >25% cellularity), an absolute neutrophil count >1.5 × 10³/µL, a platelet count >100 × 10³/µL, and resolution of all extramedullary disease, complete remission (CR) was achieved in 56 patients (93.3%), while the remaining patients experienced treatment failure with >5% blasts cells. Initial risk stratification was performed based on age, WBC count at diagnosis, blast cell counts at day 8, and key cytogenetic alterations, including BCR::ABL1 and ETV6::RUNX1 fusions, MLL rearrangements, hyperdiploidy, and hypodiploidy. For pediatric cases, based on these criteria, 8 cases were classified as high risk (HR) and the remaining 32 cases as standard risk (SR). Among adult patients, 19 were assigned to the high-risk group (Table 1).
DNA extraction
Blood and bone marrow (BM) samples were digested in a proteinase K lysis buffer containing 100 μg/mL proteinase K, 5 mM NaCl, 50 mM Tris–HCl (pH 7.5), 0.5% SDS, and 10 mM EDTA, then incubated at 56 °C for 2–4 hours with constant agitation. After digestion, DNA was extracted with phenol/chloroform/isoamyl alcohol mixture (25:24:1) and precipitated with absolute ethanol. The DNA pellet was collected, washed with 70% ethanol, air-dried, and rehydrated in sterile water. The quality and quantity of the extracted DNA were assessed with a NanoDrop® 2000 spectrophotometer (Thermo Scientific). DNA samples were stored at −20 °C until used for downstream PCR and MLPA analyses.
MLPA analysis
To identify CNAs in ALL samples, MLPA was performed using the SALSA P335-C2 kit (MRC Holland, Amsterdam, Netherlands), following the manufacturer’s instructions. The P335 probe mix allows for the detection of deletions and duplications in genes involved in lymphocyte differentiation and cell cycle regulation (IKZF1, CDKN2A/B, PAX5, EBF1, ETV6, BTG1, and RB1), as well as in genes located in the PAR1 of chromosomes X and Y (CRLF2, CSF2RA, SHOX, IL3RA, and P2RY8). These target genes and chromosomal regions are known to play an important diagnostic or prognostic role in ALL. They were selected after a thorough literature review and based on recommendations from experts in ALL research. The probemix also contains 13 reference probes serving as internal controls for data normalization. In addition to the patient samples, ten healthy control DNA samples were included for inter-sample normalization. MLPA steps, including DNA denaturation, probe hybridization, ligation, and PCR amplification, were performed in a 96-well PCR thermocycler (BioGener Thermal Cycler). PCR products were separated by capillary electrophoresis on an ABI 3500 Prism Genetic Analyzer (Applied Biosystems). MLPA data were analyzed using Coffalyser.Net™ software (MRC Holland) with default settings, which allowed for intra-sample and inter-sample normalization using the manufacturer’s internal reference probes and the healthy control samples, respectively. Peak heights from each sample were compared to those of healthy donor controls to calculate relative ratio values. A cut-off ratio of ≥ 1.3 was used to define duplications, while a ratio of ≤ 0.75 indicated deletions. Alterations in CDKN2A and/or CDKN2B were combined and reported as CDKN2A/2B abnormalities. Similarly, an alteration in at least one gene in the PAR1 region (CRLF2, CSF2RA, IL3RA, or SHOX) was classified as a PAR1 abnormality. Additionally, the IKZF1^plus^ profile was defined based on the report of stanulla. et al [31]. Briefly, the patient was considered IKZF1^plus^ positive if deletions of the IKZF1 gene coexisted with deletions of CDKN2A/CDKN2B, PAX5, and/or PAR1 region.
Multiplex PCR for ERG gene deletions
To further investigate cases meeting the IKZF1^plus^ profile criteria, and as ERG has been shown to mitigate the poor prognosis associated with IKZF1 deletions, ERG gene deletion analysis was performed by multiplex PCR, following previously established primers and PCR conditions [32]. As an internal control, an additional forward primer located in exon 11 of the ERG gene (ERG11F: cagagggctccttcaaacacag) was included in the PCR reactions, which generate a 620 bp control fragment [32].
Statistical analyses
The statistical analyses focused exclusively on gene deletions; gene duplications were excluded due to their low frequency, except for the PAR1 region, where only duplications were considered. Data were analyzed using SPSS software (version 22.0). Differences between categorical variables were compared using the chi-square test or Fisher’s exact test, as appropriate. Survival distributions were analyzed using the Kaplan–Meier method and compared using the log-rank test. Overall survival (OS) was calculated from the date of diagnosis to death or last follow-up. Relapse-free survival (RFS) was calculated from CR to the date of first relapse. The Cox proportional hazards model was used to obtain the estimate and the 95% confidence interval (CI) of the hazard ratio (HR) of the instantaneous event rate in one group versus another. Univariate and multivariate Cox hazard models were used to determine independent prognostic predictors. Multivariable analyses were performed to investigate the independent impact of IKZF1 on OS and RFS using Cox regression models, which are based on age (≥ vs. < 30 years), BCR::ABL1 (presence vs. absence), diploid karyotype (hyperdiploïdy vs. other status), WBC (≥ vs. < 50.10^3^/µl), MRD day-33 (≥ vs. < 10^-2^), MRD day-63 (≥ vs. < 10^-3^), Initial risk stratification and treatment protocol (GRALL-2014 trial vs. EORTC-CLG 58951 trial). Variables associated with p < 0.05 were considered statistically significant. CNA status was analyzed dichotomously as the deleted versus non-deleted (excluding duplication), without distinction between different isoforms.
Results
Copy number alteration frequencies in Tunisian ALL cases
A total of 130 CNAs, including intragenic deletions (n = 89) and duplications (n = 51), were identified in 42 out of 60 (70%) patients with ALL. Notably, 71.4% of these cases (n = 30) had at least two combined CNAs. Deletions were generally more frequent than duplications (72% vs. 28%), except in genes within the PAR1 region, which were predominantly duplicated (16.6%, n = 10/60). The most frequently detected deletions, either alone or in combination, concerned the CDKN2A/2B (33.3%, n = 20), IKZF1 (30%, n = 18), and PAX5 genes (25%, n = 15). Focusing on the spectrum of IKZF1 gene alterations, whole-gene deletions were mainly seen in 8 of 18 deleted cases (44.4%), followed by the ∆4–7 deletion (IK6 variant), which produces the dominant-negative isoform (n = 5; 29.4%). The ∆2–7 and ∆4–8 deletions were each found in 2 cases (11.7%), while the ∆2–8 deletion was present in only 1 case (5.8%), illustrating the diversity of IKZF1 deletion profiles. Regarding the duplication in the PAR1 region, whole-gene duplication was observed in 6 cases, while 4 patients harbored partial duplications, mainly affecting the CSF2RA gene.
Since ERG gene deletions were not detected in any of the 60 analyzed cases, the co-occurrence of IKZF1 deletions with deletions in PAX5, CDKN2A/2B, and/or PAR1 allowed the identification of an IKZF1^plus^ profile, as defined by Stanulla et al. (2018), in 10 out of the 18 patients with IKZF1 deletions (55.5%). It was detected in co-occurrence with PAX5 and CDKN2A/2B deletions in 5 cases, with PAX5 deletions alone in 3 cases, and with CDKN2A/2B deletions alone in the remaining 2 cases.
CNA frequencies according to baseline characteristics
The presence or absence of recurrent CNAs, including deletions in IKZF1, PAX5, CDKN2A/2B, JAK2, and BTG; duplications in the PAR1 region; as well as the IKZF1^plus^ profile, was assessed based on the baseline characteristics of Tunisian ALL patients (Fig 1). Our analysis revealed that IKZF1 deletions and the IKZF1^plus^ profile, defined by the co-occurrence of IKZF1 deletions with deletions in genes such as PAX5 or CDKN2A/2B, were significantly associated with adult ALL patients (p = 0.034 and p = 0.012, respectively) and with a high WBC count at diagnosis (p = 0.023 for both), as well as the presence of a BCR::ABL1 translocation (p < 0.001 and p = 0.021, respectively). Furthermore, BTG deletions were significantly associated with female gender (p = 0.007), while PAR1 region duplications were significantly associated with hyperdiploïdy (p = 0.008) (Table 2).
Table 2: CNA status according to patient characteristics and response to treatment in the entire group (n = 60).
Oncoprint illustrating the molecular characterization of the CNA spectrum across ALL subtypes and clinical parameters in Tunisian patients (n = 60).Oncoplot presents the landscape of CNAs detected by the MLPA P335 panel, alongside corresponding clinical features for each patient.
When analyzing the B-ALL subtype alone, these correlations were further confirmed. Moreover, other associations were observed, particularly the frequency of PAR1 region duplications and JAK2 gene deletions with pediatric/young adult cases (p = 0.06 and p = 0.05, respectively), as well as the significant association of JAK2 gene deletions with BCR::ABL1-positive cases (p = 0.016) (S1 Table). Regarding the T-ALL group, no significant associations were observed with the previous baseline characteristics. In contrast, comparison of CNAs between B-ALL and T-ALL cases revealed a significantly higher frequency of RB1 gene deletions in the T-ALL group (p = 0.011).
To further characterize the genomic landscape within each age subgroup, our analysis revealed that IKZF1 deletions remained significantly associated with the BCR::ABL1 translocation in both the pediatric/young adult and adult subgroups (p = 0.021 and p = 0.020, respectively). Age-specific associations were also identified, most notably the significant association between duplications in the PAR1 region and a hyperdiploid karyotype, observed exclusively among pediatric/young adult patients (p = 0.008), (S2 and S3 Tables).
CNAs’ status correlation with primary treatment response
To further investigate the clinical relevance of CNAs, we analyzed their association with primary treatment response, including corticosteroid response at day 8 and MRD status at days 33 and 63. Among the CNAs examined, IKZF1 deletions showed a significant association with corticosteroid resistance, particularly within the B-ALL subgroup (p = 0.037) and among pediatric/young adult patients (p = 0.025). In the overall cohort, deletions affecting this key hematopoietic regulatory factor were also consistently associated with positive MRD during the induction phase on both day 33 and day 63 (p = 0.024 and p < 0.001). Additional significant associations were identified, particularly those involving alterations that co-occur with IKZF1 deletions, PAX5 and CDKN2A/2B gene deletions which define the IKZF1^plus^ profile. Similar trends were observed in subgroup analyses, particularly among adult patients and within the B-ALL subtype (Table 2, S1-S3 Tables).
CNAs’ survival rates and outcomes
The impact of gene deletions on survival, taken separately and in combined within the IKZF1plus profile was assessed using the Kaplan–Meier method. We showed that IKZF1 and PAX5 deletions were significantly associated with poor survival outcomes in the overall cohort. Notably, patients with IKZF1 deletions had significantly lower outcomes in both RFS and OS than non-deletion carriers (p < 0.001). Additionally, patients with CDKN2A/2B deletions had shorter RFS than patients with the wild-type form (p = 0.006). Importantly, cases classified according to the IKZF1^plus^ profile also exhibit significantly poor survival outcomes with short OS and RFS rates (p < 0.0001), thus reinforcing its prognostic relevance (Fig 2).
Overall survival and cumulative incidence of relapse in Tunisian ALL patients according to CNA status (deletion vs. no deletion).Red Kaplan–Meier curves represent patients harboring specific gene deletions, while blue curves correspond to wild-type cases. OS: Overall survival, RFS: relapse free survival.
When evaluating survival outcomes within each subgroup, these associations remained statistically significant in the B-ALL subtype, where JAK2 deletions were also correlated with shorter OS (p = 0.021) and reduced RFS (p = 0.028) (S1 Fig). Furthermore, patients carrying IKZF1 deletions or the IKZF1^plus^ profile consistently showed poorer OS and RFS in both pediatric/young adult and adult groups. Deletions affecting PAX5 and CDKN2A/2B were likewise associated with inferior survival (p = 0.005 and p = 0.021, respectively), especially in adult patients (S2 and S3 Figs).
To assess whether IKZF1 deletions and the IKZF1^plus^ profile retained prognostic significance independent of other clinical and molecular variables, a multivariate Cox regression model was performed. The model included the following covariates: age, BCR::ABL1 translocation status, diploidy, WBC count at diagnosis, MRD status at days 33 and 63, and treatment protocols. Our results demonstrate that IKZF1 deletions are independently associated with a significantly increased risk of death. In the overall cohort, patients harboring these deletions had a markedly worse OS (HR = 3.51; p = 0.027), with the effect being even more pronounced in the B-ALL subtype (HR = 6.66; p = 0.009) and in adult patients (HR = 9.16; p = 0.01). These findings highlight IKZF1 deletions as a robust independent predictor of poor prognosis. Notably, MRD levels at days 33 and/or 63 remained statistically significant predictors of survival, underscoring their continued value as a key prognostic marker (Table 3, S4 and S6 Tables). In contrast, the IKZF1^plus^ profile was significantly associated with OS and RFS in univariate analyses, these associations were not maintained in multivariate models, suggesting that its prognostic impact may be influenced by other factors.
Table 3: Multivariate Cox model assessing the impact of IKZF1 deletions on survival in ALL patients (n = 60).
Risk stratification refinement based on the IKZF1 gene status
To further investigate the crucial prognostic value of IKZF1 deletions, we analyzed their combination with established prognostic factors; Model 1: including initial risk stratification and Model 2: in combination with MRD status, assessing their impact on OS, RFS, and CIR. Based on this integrated approach, all patients in our cohort were reclassified into three molecular risk groups by combining IKZF1 status with initial risk stratification: Standard molecular risk (initial standard-risk and wild-type IKZF1), High molecular risk (initial high-risk and wild-type IKZF1) and Very high molecular risk (initial standard- or high-risk with IKZF1 deletion).
This redefined classification revealed a significant prognostic impact, with patients in the very high molecular risk group having significantly lower 5-year OS rates (10.5% vs. 61.5% vs. 96.4%, p < 0.001), compared with the high and standard molecular risk groups. There was also a statistically significant difference in OS between standard-risk with IKZF1 deletion and cases in the same groups with wild-type IKZF1 gene (96% vs. 21%, p < 0.001). These findings highlight the critical importance of incorporating molecular profiling, particularly early IKZF1 deletion status, into risk stratification to improve prognostic accuracy and guide more effective therapeutic decision-making.
A second redefined model recently described by Deng et al. (2025) was based on MRD status and IKZF1 gene profile, stratifying patients into Standard (MRD-negative and IKZF1 wild-type), intermediate (MRD-positive or IKZF1 deletion), and high-risk (MRD-positive and IKZF1 deletion) groups. Similarly, the standard risk group was strongly associated with higher 5-year OS rates than the intermediate and high-risk groups. This result confirmed the low impact of the IKZF1 gene deletion rather within the standard risk group (Fig 3). No statistically significant results were noted for the RFS level.
Overall survival of ALL patients according to the new risk stratification (n = 60).Patients were stratified into Model 1: Standard molecular risk (initial standard-risk and wild-type IKZF1), High molecular risk (initial high-risk and wild-type IKZF1) and Very high molecular risk (initial standard- or high-risk with IKZF1 deletion); Model 2: Standard (MRD-negative and IKZF1 wild-type), intermediate (MRD-positive or IKZF1 deletion), and high-risk (MRD-positive and IKZF1 deletion) groups.
Discussion
Our study is the first to assess the frequency, heterogeneity, and clinical significance of CNAs in an unselected cohort of both pediatric/young adult and adult ALL Tunisian patients. Although CNAs are well established as essential prognostic markers in contemporary treatment protocols, they are not routinely assessed in our hospital settings [5,8,33,34]. Therefore, this study was designed to characterize the CNAs profile in the Tunisian ALL population, to identify specific genetic signatures that could guide future risk-adapted therapeutic strategies and ultimately enhance clinical outcomes.
To achieve this aim, we used the MLPA technique, a reliable and efficient molecular method capable of detecting a broad spectrum of CNAs, including IKZF1 deletions and other common gene abnormalities in ALL. MLPA offers a practical and scalable approach for large-scale clinical screening and research, making it particularly suitable for implementation even in resource-limited settings, thus improving accessibility to essential genetic diagnostics. In our analysis, 70% of the ALL cases studied had genetic abnormalities, with the most frequent deletions observed in CDKN2A/2B (33.3%), IKZF1 (30%), and PAX5 (25%), while deletions in EBF1 (6.7%) and ETV6 (10%) were comparatively less frequent. Overall, the CNA distribution is consistent with previous results established among children and adult patients using the MLPA method, highlighting the increasing clinical relevance of CNA profiling in patient management. However, a literature review reveals considerable variability in reported CNA frequencies, likely due to differences in study design, particularly the selective inclusion of specific entities, such as pediatric versus adult-onset, and T-ALL versus B-ALL subtypes [4,34–35]. A major conclusion drawn from these observations is that ALL patients exhibit distinct genetic landscapes, reflecting fundamental biological differences that significantly influence clinical outcomes and therapeutic responses [36–39]. In fact, our study revealed a notable age-related variation in IKZF1 gene deletions, with a significantly higher frequency observed in adult patients (50%) compared to pediatric cases (20%). Furthermore, we observed that IKZF1 gene deletions were more frequent in patients with BCR::ABL1 translocation in the B-ALL subtype (p = 0.001). Our findings are consistent with previously reported trends, reinforcing the role of IKZF1 deletions, which often co-occur with high-risk factors such as BCR::ABL1 transcript, in contributing to increased disease aggressiveness, particularly among adult B-cell patients [36,40–43]. Compared with adult B-ALL patients, pediatric/young adult cases had a lower frequency of deletions and a higher incidence of duplications, especially in genes within the PAR1 region. Moreover, these gene duplications were significantly associated with hyperdiploïd karyotype, a cytogenetic feature historically linked to favorable prognosis in ALL [44,45]. Among these genes, the CRLF2 gene has received a particular attention due to its recurrent association with genetic abnormalities, leading to elevated expression that appears to define a distinct subgroup of B-ALL and offers potential molecular targets for treatment [46–48]. Although these abnormalities often include P2RY8::CRLF2 fusions resulting from interstitial deletions, our results revealed no such rearrangements [26]. Instead, we identified the presence of additional copies of the CRLF2 locus in 9 cases, probably due to supernumerary X chromosomes, which could also contribute to CRLF2 overexpression [24,49,50]. Even though duplications in the PAR1 region are not frequently documented, similar findings have been reported by Schmäh et al. (2017). They showed that high CRLF2 expression, associated with increased gene copy number, is typically characterized by rare additional deletions and hyperdiploidy karyotypes, in contrast to P2RY8::CRLF2 fusion and IGH::CRLF2 translocation cases, which are associated with additional deletions. Regarding the CNA frequencies within the B and T immunophenotypic groups, our results revealed uneven distributions, particularly among RB1 gene deletions, which were significantly more frequent in T-ALL cases, aligning with previous findings [51]. Although RB1 is traditionally considered to play a crucial role in B-cell differentiation, emerging data suggest that its functional impact varies depending on the hematopoietic context. Indeed, most studies have associated RB1 deletions with impaired differentiation and increased aggressiveness in B-lineage leukemia [52,53].
The second major challenge of our study was to assess the importance of the identification of CNAs, often described as a poor prognostic factor, in the treatment response and survival of Tunisian patients with T/B precursor ALL. In the early stages of treatment, glucocorticoid resistance represents a critical factor in ALL and is recognized as a major contributor to therapeutic failure and disease relapse. The resistant phenotype has been linked to multiple molecular drivers, including activation of the AKT and ERK signaling pathways, which are frequently triggered in IKZF1-deficient leukemic cells [54]. Furthermore, combined loss of BTG1 and IKZF1 has been shown to further increase glucocorticoid resistance [47]. In our cohort, all 16 ALL patients with a resistant phenotype had at least one CNA in IKZF1, CDKN2A/2B, and/or PAX5 genes, with a statistically significant association observed specifically for IKZF1 gene deletions, particularly among B-cell ALL cases (p = 0.037). Similar findings were reported by Braun et al. (2022) [55], who studied 373 children with B-cell precursor ALL and observed significantly lower glucocorticoid responses in patients with IKZF1 deletions. Although the role of CDKN2A/2B deletions is still under investigation, other studies have concluded that patients with CDKN2A/B deletions were more frequently steroid-resistant and exhibited a higher risk of poor prednisolone response [22]. Additionally, these gene deletions have been shown to correlate with MRD positivity during various treatment assessments [22,56,57]. Consistent with these findings, our cohort also demonstrated a significant association between IKZF1 deletions and MRD positivity, specifically on day-33 of induction therapy and day-63 following consolidation, in the overall ALL patients (p = 0.024 and p < 0.001, respectively), as well as within the B-ALL subtype (p = 0.035 and p = 0.002, respectively) and among adult patients (p = 0.005 and 0.003, respectively).
As expected, our findings showed that CNAs associated with primary treatment failure are also powerful prognostic markers, closely related to poor outcomes, as indicated by a significant reduction in OS as well as increased CIR. Interestingly, deletions in the transcription factor IKZF1 have been widely described as an independent marker of poor prognosis in both pediatric and adult ALL [39,42,43]. Consistent with these data, our study showed that the presence of IKZF1 gene deletions is a reliable and independent predictor of poor survival outcomes in ALL. This association was confirmed by multivariate analyses, revealing that patients harboring IKZF1 deletions had a markedly worse OS in the overall cohort (HR = 3.51; p = 0.027), with an even stronger adverse impact observed in the B-ALL subtype (HR = 6.66; p = 0.009) and in adult patients (HR = 9.16; p = 0.01). Our results closely mirror previous research, which has consistently shown that IKZF1 deletions are linked to inferior survival outcomes and an increased risk of relapse [39,45,58–60], underscoring the critical importance of IKZF1 status in risk stratification and therapeutic decision-making in ALL.
To improve outcome prediction and refine risk assessment in ALL, the integration of IKZF1 deletions into established prognostic factors, such as initial risk stratification and MRD, is increasingly proposed. This approach allows the identification of subgroups with variable outcomes, as IKZF1 deletions are associated with poor prognosis even within the standard-risk group. Therefore, these patients can be considered for treatment intensification or alternative therapeutic strategies. Notably, studies by Waanders et al. (2011) and Deng et al. (2025) have demonstrated that the combination of MRD status and IKZF1 deletions offers superior prognostic value than either factor alone. In line with these findings, our results showed that the highest 5-year OS rate was observed in patients with both MRD-negative status and wild-type IKZF1, reaching 96%. This rate was significantly higher than in patients with either negative MRD alone (70%) or IKZF1 wild-type alone (78%). In addition, a redefined model based on initial risk and IKZF1 deletion status revealed a significant prognostic impact, as patients in the very high molecular risk group exhibited markedly inferior 5-year OS rates, compared with high and standard molecular risk (p < 0.001), consistent with previous studies [43,61]. Such a refined approach to risk assessment could enable more personalized and effective treatment strategies for ALL, potentially reducing treatment-related toxicity and improving long-term outcomes.
The relatively small sample size decreases statistical power and may limit the generalizability of our findings. Additionally, the absence of complementary cytogenetic analyses and comprehensive molecular profiling restricts the full characterization of the genetic landscape and its clinical significance. The dataset’s limitations also limit the application of advanced statistical frameworks, such as the Moorman classification, which could offer deeper insights into the patterns and prognostic implications of concomitant CNAs. Therefore, future studies with larger, well-characterized cohorts and integrated genomic approaches are crucial to approve these results.
Conclusion
Our study highlights the clinical importance of early detection of CNAs, including IKZF1 deletion, using MLPA. Integrating this molecular approach into routine diagnostics should refine risk stratification, enable more accurate diagnosis, and guide treatment decisions. By identifying genetic alteration profiles early, clinicians can more effectively tailor therapeutic strategies, potentially improving patient outcomes.
Supporting information
S1 TableCorrelation of CNA frequencies with clinicopathological features and treatment responses in the B-ALL group (n = 45).(DOCX)
S2 TableCorrelation of CNA frequencies with clinicopathological features and treatment responses in the ALL pediatric/young adult group (n = 40).(DOCX)
S3 TableCorrelation of CNA frequencies with clinicopathological features and treatment responses in the ALL Adults group (n = 20).(DOCX)
S4 TableMultivariate Cox model assessing the impact of IKZF1 deletions on survival in B-ALL cases (n = 45).(DOCX)
S5 TableMultivariate Cox model assessing the impact of IKZF1 deletions on survival in the ALL pediatric/young adult group (n = 40).(DOCX)
S6 TableMultivariate Cox model assessing the impact of IKZF1 deletions on survival in the ALL adult group (n = 20).(DOCX)
S1 FigOverall survival and cumulative incidence of relapse in the B-ALL subtype according to CNA status (deletion vs. no deletion).Red Kaplan–Meier curves indicate patients exhibiting CNA-associated gene deletions; blue curves represent wild-type (non-deleted) genotypes. OS: Overall survival, RFS: relapse free survival.(TIF)
S2 FigOverall survival and cumulative incidence of relapse in the Pediatric/Young adult ALL cases according to CNA status (deletion vs. wild-type).Kaplan–Meier curves illustrate OS and CIR stratified by the presence or absence of specific gene deletions. Red curves represent patients harboring CNA-associated deletions, whereas blue curves correspond to wild-type (non-deleted) cases. OS: Overall survival, RFS: relapse free survival.(TIF)
S3 FigOverall survival and cumulative incidence of relapse in adult ALL cases according to CNA status (deletion vs. wild-type).Kaplan–Meier curves correspond to OS and CIR stratified by the presence or absence of specific gene deletions in each adult case. Red curves represent patients harboring CNA-associated deletions, whereas blue curves correspond to wild-type (non-deleted) cases. OS: Overall survival, RFS: relapse free survival.(TIF)
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Puckett Y, Chan O. Acute lymphocytic leukemia. Treasure Island (FL): Stat Pearls Publishing; 2025.29083572 · pubmed ↗
- 2Roberts KG. Genetics and prognosis of ALL in children vs adults. Hematology Am Soc Hematol Educ Program. 2018;2018(1):137–45. doi: 10.1182/asheducation-2018.1.137 30504302 PMC 6245970 · doi ↗ · pubmed ↗
- 3Terwilliger T, Abdul-Hay M. Acute lymphoblastic leukemia: a comprehensive review and 2017 update. Blood Cancer J. 2017;7(6):e 577. doi: 10.1038/bcj.2017.53 28665419 PMC 5520400 · doi ↗ · pubmed ↗
- 4Crepinsek K, Marinsek G, Kavcic M, Prelog T, Kitanovski L, Jazbec J, et al. Clinical impacts of copy number variations in B-cell differentiation and cell cycle control genes in pediatric B-cell acute lymphoblastic leukemia: a single centre experience. Radiol Oncol. 2021;56(1):92–101. doi: 10.2478/raon-2021-0050 34957727 PMC 8884847 · doi ↗ · pubmed ↗
- 5Yoon J-H, Kwag D, Min GJ, Park S-S, Park S, Lee S-E, et al. Adverse Prognostic Role of Copy Number Alterations and mutations in adults with philadelphia chromosome-negative Acute Lymphoblastic Leukemia. Blood. 2023;142(Supplement 1):4342–4342. doi: 10.1182/blood-2023-191174 · doi ↗
- 6Yi M, Zhou L, Li A, Luo S, Wu K. Global burden and trend of acute lymphoblastic leukemia from 1990 to 2017. Aging (Albany NY). 2020;12(22):22869–91. doi: 10.18632/aging.103982 33203796 PMC 7746341 · doi ↗ · pubmed ↗
- 7Hu Y, Zhang X, Zhang A, Hou Y, Liu Y, Li Q, et al. Global Burden and Attributable Risk Factors of Acute Lymphoblastic Leukemia in 204 Countries and Territories from 1990-2019: Estimation Based on Global Burden of Disease Study 2019. Springer Science and Business Media LLC. 2021. doi: 10.21203/rs.3.rs-508326/v 134664286 · doi ↗ · pubmed ↗
- 8Besbes S, Hamadou WS, Boulland ML, Youssef YB, Achour B, Regaieg H, et al. Minimal residual disease detection in Tunisian B-acute lymphoblastic leukemia based on immunoglobulin gene rearrangements. Braz J Med Biol Res. 2017;50(1):e 5426. doi: 10.1590/1414-431X 20165426 28099581 PMC 5264541 · doi ↗ · pubmed ↗