Identification of a Chinese patient with MTSS2-causing intellectual disability by WES reanalysis: a case report
Duan Li, Wen Zhang, Huifen Mei, Xiaodan Chen, Zhikun Lu, Pengfei Wu, Li Liu, Yunting Lin

TL;DR
A Chinese patient with intellectual disability and distinctive facial features was found to have a rare MTSS2 gene variant through reanalysis of genetic data.
Contribution
This study reports a new patient with IDDOF in China and highlights the importance of reanalyzing WES data for improved diagnosis.
Findings
Reanalysis of WES data identified a de novo MTSS2 variant (c.2011C>T) in a patient with IDDOF.
The c.2011C>T variant in MTSS2 is suggested as a hotspot for causing IDDOF.
WES reanalysis improved diagnostic yield in a previously negative case.
Abstract
Intellectual developmental disorder with ocular anomalies and distinctive facial features (IDDOF) is an extremely rare disease caused by a heterozygous pathogenic variant in the MTSS2 gene with an autosomal dominant inheritance pattern. To date, only 10 patients with IDDOF and one pathogenic variant in the MTSS2 gene have been reported. Here, we present a new Chinese patient with IDDOF, who is the 11th patient worldwide and the second case in China. The proband presented with relative microcephaly, distinctive facial features of bitemporal narrowing and ptosis, ophthalmological and auditory findings, hypotonia, psychomotor developmental delay, intellectual disability, and emotional and behavioral problems. Whole exome sequencing (WES) initially did not find a phenotype-contributing variant in 2021, whereas reanalysis of WES data in 2024 revealed that the de novo c.2011C>T(p.Arg671Trp)…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
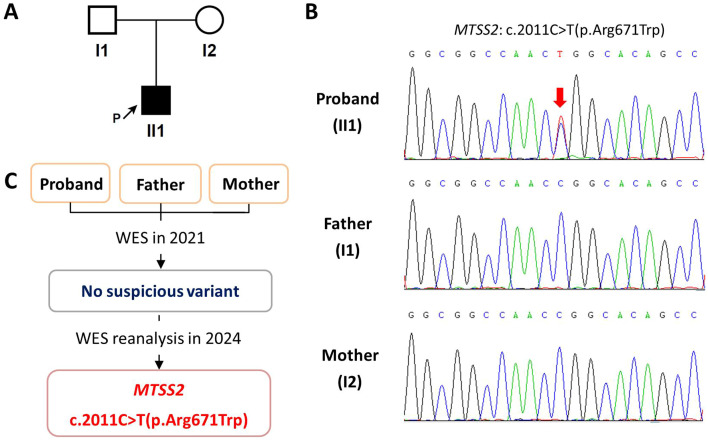 Figure 1
Figure 1| Family model | Gene | Chr | Transcript | Variant | Allele | Inheritance | OMIM-disease |
|---|---|---|---|---|---|---|---|
| Dominant |
| 16 | c.2011C>T(p.Arg671Trp) | Het |
| – | |
| Recessive |
| X | c.3422G>A(p.Arg1141Gln) | Hem | Maternal | Shukla-Vernon syndrome (XLR) | |
| Recessive |
| X | c.1793A>T(p.Glu598Val) | Hem | Maternal | Hypothyroidism, central, and testicular enlargement (XLR) | |
| Compound heterozygous |
| 14 | c.5531C>T(p.Ala1844Val) | Het | Paternal | – |
| Variant | PROVEAN | SIFT | PolyPhen-2 | Mutation taster | FATHMM |
|---|---|---|---|---|---|
| Neutral | Tolerated | Possibly damaging/benign | Benign | Tolerated | |
| Neutral | Tolerated | Possibly damaging/possibly damaging | Benign | Tolerated |
| Patient | P1 | P2 | P3 | P4 | P5 | P6 | P7 | P8 | P9 | P10 | P11 | Mean ± SD or percentage |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reference | Huang Y et al. ( | Corona-Rivera JR et al. ( | Dominicis A et al. ( | This study | / | |||||||
| Gender | Male | Female | Male | Male | Male | Male | Male | Female | Male | Male | Male | Male:female = 9:2 |
| Ethnicity | European | UK | Dutch | European | Chinese | Mexican | European | European | European | European | Chinese | / |
| Age (years) | 8 | 42 | 15 | 1.17 | 1.75 | 7 | 10 | 21 | 7.67 | 12 | 5.25 | 8.8 ± 5.7 |
| Growth retardation | − | − | Unknown | − | − | + | − | + | − | − | − | 20.0%(2/10) |
| Facial dysmorphism | + | + | + | + | Unknown | + | + | + | + | + | + | 100.0%(10/10) |
| Microcephaly or relative microcephaly | + | + | + | + | + | + | + | + | + | + | + | 100.0%(11/11) |
| Ophthalmological anomalies | + | + | + | + | + | + | + | − | + | − | + | 81.8%(9/11) |
| Hearing impairment | + | + | Unknown | − | − | Unknown | − | + | − | − | + | 44.4%(4/9) |
| ID | + | + | + | + | + | + | + | + | + | + | + | 100.0%(11/11) |
| Autism spectrum disorder | + | − | + | Not applicable | Not applicable | + | − | − | − | − | − | 33.3%(3/9) |
| Seizures | − | + | − | − | − | + | + | + | − | − | − | 36.7%(4/11) |
| Gene variant | / | |||||||||||
| Inheritance |
|
|
|
|
| Unknown (absence in his co-twin and mother) |
|
|
| Unknown |
| / |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenomics and Rare Diseases · Ocular Disorders and Treatments · Genomic variations and chromosomal abnormalities
Introduction
1
Intellectual disability (ID) is one of the most prevalent neurodevelopmental disorders affecting 1%-3% of the general population (1). To date, more than 2,000 genes have been implicated in ID with high genetic heterogeneity and phenotypic variability. Therein, de novo variants with autosomal dominant trait are prominent contributors to ID in non-consanguineous families (2).
The MTSS2 gene (OMIM * 615951), formerly known as MTSS1L gene, encodes MTSS I-BAR domain-containing protein 2, which is mainly expressed in the central nervous system (3). In 2022, a recurrent de novo c.2011C>T(p.Arg671Trp) variant in the MTSS2 gene (NM_138383.3) was revealed to cause a novel ID syndrome in five unrelated individuals through a dominant-negative mechanism (4). This associated disorder is now named “Intellectual developmental disorder with ocular anomalies and distinctive facial features (IDDOF)” (OMIM # 620086) in the Online Mendelian Inheritance in Man (OMIM) database. Subsequently, an additional case of IDDOF was reported in 2023 (5). Recently, four new cases have been added to the literature (6). However, the mutational spectrum of this disorder has not expanded, since all 10 reported patients share the same MTSS2 c.2011C>T(p.Arg671Trp) variant.
Here, we present a new Chinese boy with IDDOF, who is the 11th patient worldwide and the second case in China, and describe his diagnostic process.
Case presentation
2
The proband (II1) is the first child of the non-consanguineous healthy parents (I1 and I2) (Figure 1A). He was delivered naturally at term with a normal length of 51 cm (0.3 SD) and weight of 3.6 kg (0.7 SD) (7). There was no history of prenatal issues or birth injury. After birth, the proband was noticed to have ptosis and hypotonia and gradually developed psychomotor delay. He achieved head control at 4 months of age, could turn over at 8 months of age, started walking independently at 1 year and 10 months of age, and enabled to speak until 2 years and 7 months of age. His brain magnetic resonance imaging at 7 months of age and electroencephalogram at 2 years and 2 months of age had no obvious abnormality. His skeletal X-ray at 1 year and 6 months of age indicated spina bifida occulta of the fifth lumbar vertebra, and auditory evoked potential at 2 years and 3 months of age showed a slight increase of bilateral hearing thresholds.
The family pedigree and molecular findings of the enrolled family. (A) The family pedigree. (B) Sanger sequencing chromatograms of the enrolled family. The red arrow indicates the variant. (C) The molecular diagnostic process of the proband.
The proband first presented to our clinic at 2 years and 7 months of age in 2021 because of ID, speech delay, and motor incoordination. He presented with distinctive facial features of bitemporal narrowing and ptosis. He had a normal height of 93.2 cm (−0.2 SD) and a weight of 14.5 kg (0.4 SD) but relative microcephaly with an occipitofrontal circumference of 47.5 cm (−1.4 SD) and chest circumference of 51 cm (0.1 SD) (7, 8). Physical examination found a small cafe-au-lait spot on his left thigh and an enlargement of his left scrotum. Ophthalmological examination revealed astigmatism and myopia. Furthermore, the proband suffered from emotional and behavioral problems, such as bad temper, irritability, and automatic speaking. No abnormality was found in his biochemical and metabolic profiles, karyotype analysis, and chromosomal microarray analysis.
To identify the phenotype-producing variant, whole exome sequencing (WES) of the proband–parent trio was performed. The analysis identified several variants: a de novo variant in the MTSS1L gene; two hemizygous variants in the BCORL1 and IGSF1 genes, both inherited from the mother; and a compound heterozygous variant in the AHNAK2 gene, inherited from each parent (Table 1). Among them, only BCORL1 and IGSF1 were known disease-causing genes, but the identified variants in these two genes were predicted to be tolerated by in silico analyses (Table 2). Therefore, none of the identified variants could explain the proband’s phenotypes.
The proband was then lost to follow-up until 5 years and 3 months of age when he revisited our clinic for further examination. He remained with a normal height and weight, and relative microcephaly with an occipitofrontal circumference of 49 cm (−1.7 SD) and chest circumference of 55 cm (−0.4 SD) (8). There was no significant improvement for his neurological phenotypes of ID, speech delay, and motor incoordination.
To further investigate if there is an underlying molecular basis, WES data were reanalyzed in 2024. Surprisingly, the de novo c.2011C>T(p.Arg671Trp) heterozygous variant of the MTSS1L gene, now known as MTSS2 gene, in the proband turned out to be a known pathogenic variant of IDDOF. Sanger sequencing subsequently confirmed the de novo status of this variant with presence in the proband and absence in his unaffected parents (Figure 1B).
Discussion
3
IDDOF is an extremely rare disease caused by a heterozygous deleterious variant in the MTSS2 gene with an autosomal dominant inheritance pattern. To date, only 10 patients with IDDOF and one pathogenic variant in the MTSS2 gene have been reported (4–6). Our case shared similar facial appearances and neurologic features, and the same c.2011C>T(p.Arg671Trp) heterozygous variant in the MTSS2 gene with these previous patients (Table 3), which further supports the MTSS2 gene as the cause of IDDOF and suggests the c.2011C>T(p.Arg671Trp) variant in the MTSS2 gene as a hotspot. Thus, our patient could be diagnosed as IDDOF clinically and genetically.
However, the confirmation of IDDOF did not change the supportive rehabilitation interventions for our patient, as there is no effective treatment for this disease. For his parents, prenatal diagnosis was suggested for future pregnancies because gonadal mosaicism could not be ruled out, even though the pathogenic MTSS2 variant was not detected in their blood samples.
In addition to the classical symptoms of IDDOF, our patient exhibited previously unreported phenotypes, including spina bifida occulta of fifth lumbar vertebra, an enlarged scrotum, and astigmatism. Among the 10 previously reported IDDOF patients, some showed ophthalmological manifestations such as foveal hypoplasia, progressive optic nerve atrophy, iris cysts, and myopia, while the patient from Mexico had bilateral inguinal hernias and bilateral cryptorchidism (5–7). Moreover, recent studies have implicated the MTSS2 protein in cell migration and spinogenesis (9) and have suggested that variants in the MTSS2 gene may contribute to the etiopathogenesis of spina bifida (10). Therefore, these findings collectively suggest that spina bifida and ophthalmological and genitourinary abnormalities are specific features of IDDOF.
In this study, our initial WES analysis in 2021 did not find the phenotype-causing variant in the proband, whereas the reanalysis in 2024 successfully identified it (Figure 1C). That is because the MTSS2 gene was first described to cause IDDOF in 2022 (4). Our initial WES analysis based on OMIM in 2021 could not recognize this novel causative gene uncovered later. This study is very similar to our previous report on Marbach–Rustad progeroid syndrome resulting from the de novo c.1436C>T(p.Ser479Phe) heterozygous variant in the LEMD2 gene (11). Our experience indicates that WES reanalysis of negative cases could improve diagnostic yield, particularly when there are previously unknown gene–disease associations.
Actually, our initial identification of the de novo c.2011C>T(p.Arg671Trp) variant in the MTSS2 gene in 2021 was earlier than the first gene-disease report in 2022. However, the lack of resources for further functional studies, coupled with limited access to genomic matchmaking platforms and inter-institutional collaboration, prevented us from identifying this new disease-causing gene.
In addition, although only the c.2011C>T(p.Arg671Trp) variant in the MTSS2 gene has been confirmed as the cause of IDDOF, 18 different variants in the MTSS2 gene have been documented in the Human Gene Mutation Database (Professional 2025.2). Among them, 13 variants are annotated with presence in patients having neurodevelopment disorders.
In fact, the MTSS2 gene was first described as a novel candidate gene for human neurogenetic disorders in 2015 as the c.1790C>T(p.Thr597Met) missense variant was found in two siblings presenting ID and segregated in their family with an autosomal recessive inheritance pattern (12). However, the clinical significance of this c.1790C>T(p.Thr597Met) variant needs to be further evaluated due to a high frequency of 1.6% recorded in the Kinh Vietnamese genetic variation database (13). Thereafter, several variants in the MTSS2 gene were found in patients affected by developmental disorder or autism, but their clinical significances were undefined (14–17). More studies are needed to determine whether there are novel inheritance pattern and more pathogenic variants for IDDOF.
Conclusions
4
Our study adds a new patient with IDDOF, who is the 11th case worldwide and the second case in China, and provides additional phenotypes to expand the phenotypic spectrum of this disease. Our finding supports the MTSS2 gene as the cause of IDDOF and indicates the c.2011C>T(p.Arg671Trp) variant in the MTSS2 gene as a hotspot. Our experience also underlines the potential of WES reanalysis to improve diagnostic efficiency, particularly in rediscovering previously unknown gene–disease associations.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Marrus N Hall L . Intellectual disability and language disorder. Child Adolesc Psychiatr Clin N Am. (2017) 26:539–54. doi: 10.1016/j.chc.2017.03.001, PMID: 28577608 PMC 5801738 · doi ↗ · pubmed ↗
- 2Li C Wang Y Zeng C Huang B Chen Y Xue C . Trio-whole exome sequencing reveals the importance of de novo variants in children with intellectual disability and developmental delay. Sci Rep. (2024) 14:27590. doi: 10.1038/s 41598-024-79431-x, PMID: 39528574 PMC 11555314 · doi ↗ · pubmed ↗
- 3Chatzi C Westbrook GL . Revisiting I-BAR proteins at central synapses. Front Neural Circuits. (2021) 15:787436. doi: 10.3389/fncir.2021.787436, PMID: 34975417 PMC 8716821 · doi ↗ · pubmed ↗
- 4Huang Y Lemire G Briere LC Liu F Wessels MW Wang X . The recurrent de novo c.2011 C>T missense variant in MTSS 2 causes syndromic intellectual disability. Am J Hum Genet. (2022) 109:1923–31. doi: 10.1016/j.ajhg.2022.08.011, PMID: 36067766 PMC 9606386 · doi ↗ · pubmed ↗
- 5Corona-Rivera JR Zenteno JC Ordoñez-Labastida V Cruz-Cruz JP Cortés-Pastrana RC Peña-Padilla C . MTSS 2-related neurodevelopmental disorder: Further delineation of the phenotype. Eur J Med Genet. (2023) 66:104826. doi: 10.1016/j.ejmg.2023.104826, PMID: 37657631 · doi ↗ · pubmed ↗
- 6Dominicis A Sparascio FP Stregapede F Terracciano A Verrigni D Lepri FR . MTSS 2-related disorder: refining the phenotype in four new cases and literature review. Am J Med Genet A. (2025) 19:e 64010. doi: 10.1002/ajmg.a.64010, PMID: 39890443 · doi ↗ · pubmed ↗
- 7Li H Ji CY Zong XN Zhang YQ . Height and weight standardized growth charts for Chinese children and adolescents aged 0 to 18 years. Zhonghua Er Ke Za Zhi. (2009) 47:487–92., PMID: 19951507 · pubmed ↗
- 8Coordinating Study Group of Nine Cities on Physical Growth and Development of Children, and Capital Institute of Pediatrics . A national survey on growth of children under 7 years of age in nine cities of China, 2005. Zhonghua Er Ke Za Zhi. (2007) 5:609–14. 18021536 · pubmed ↗