Semantic-enhanced heterogeneous graph learning for identifying ncRNAs associated with drug resistance
Hang Wei, Yuran Xie, Wenxiang Zhang, Linyang Li, Shuai Wu, Lin Gao

TL;DR
This paper introduces a new method to identify non-coding RNAs linked to drug resistance using an enhanced graph learning approach that improves accuracy and interpretability.
Contribution
The novel iNcRD-HG framework integrates heterogeneous molecular interactions and semantic context for better ncRNA-drug resistance predictions.
Findings
iNcRD-HG outperforms existing methods on benchmark datasets for ncRNA-drug resistance association prediction.
The framework captures complex contextual dependencies through relation-type-aware message passing.
It provides interpretable insights into synergistic pathways involved in drug resistance.
Abstract
Identifying non-coding RNAs (ncRNAs) associated with drug resistance is critical for elucidating molecular mechanisms underlying drug response, facilitating drug screening, and discovering novel therapeutic targets. While several graph neural network-based methods have been proposed to infer ncRNA-drug resistance associations, they remain fundamentally constrained by semantic distortion induced by a sparse bipartite network and neglect of relational semantics among molecular entities, ultimately compromising both predictive reliability and biological interpretability. In this study, we propose iNcRD-HG, a novel framework for identifying ncRNA-drug resistance associations. The framework addresses three critical aspects: constructing a context-enriched heterogeneous network that integrates six distinct molecular interaction types with bio-entity-specific attributes, developing a…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
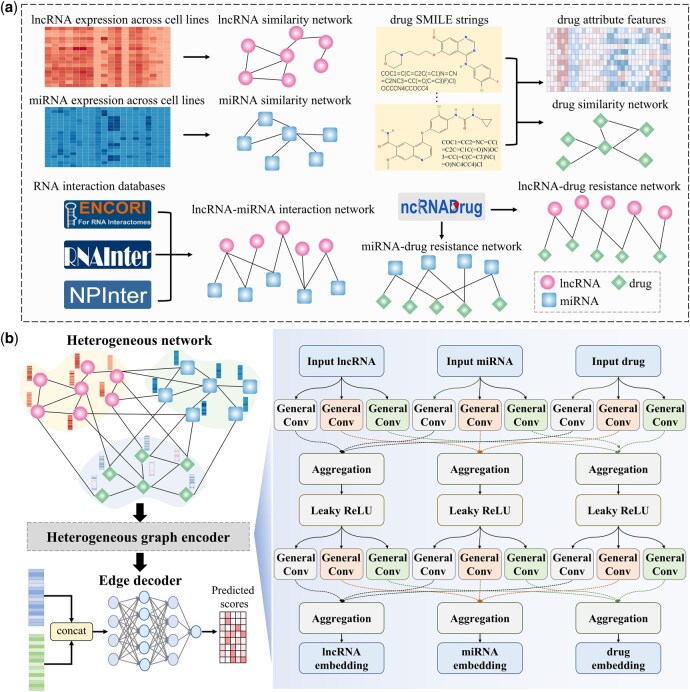 Figure 1
Figure 1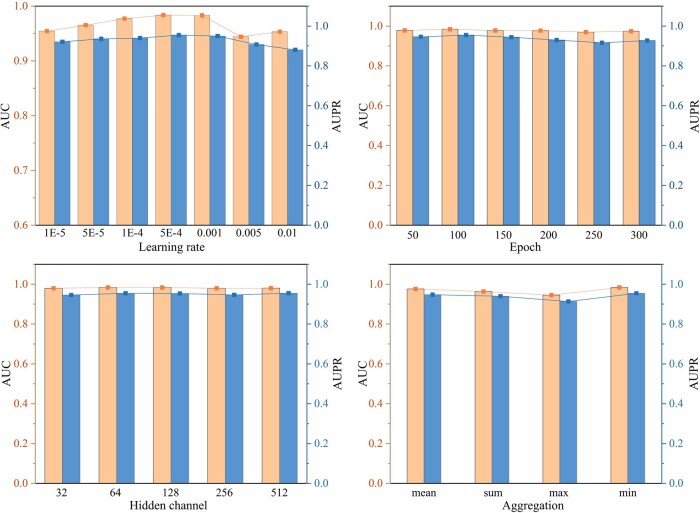 Figure 2
Figure 2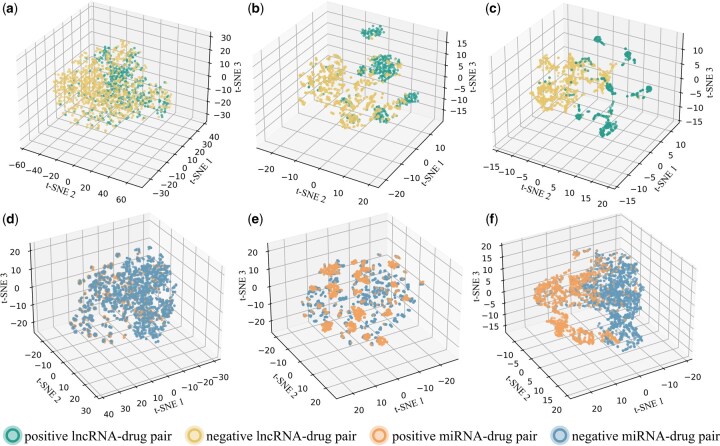 Figure 3
Figure 3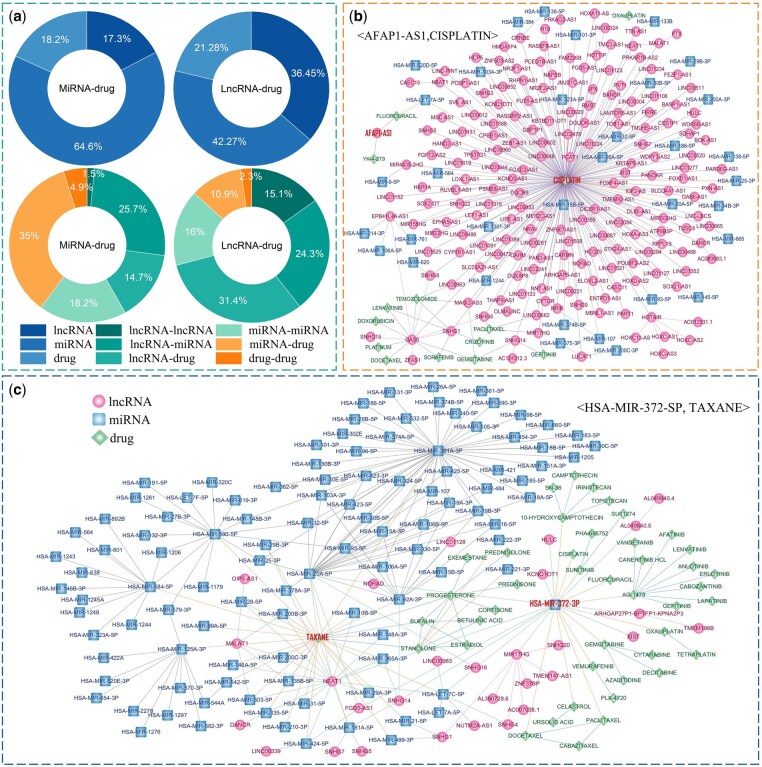 Figure 4
Figure 4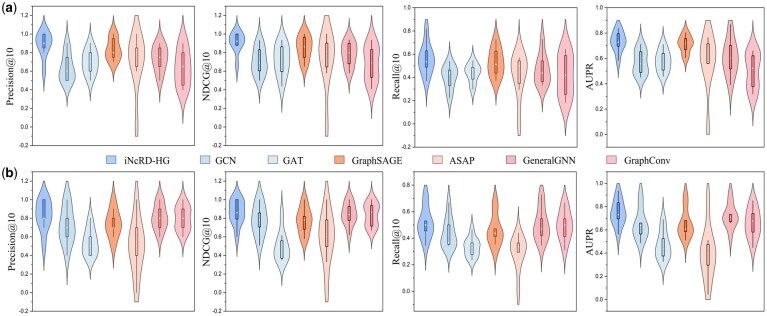 Figure 5
Figure 5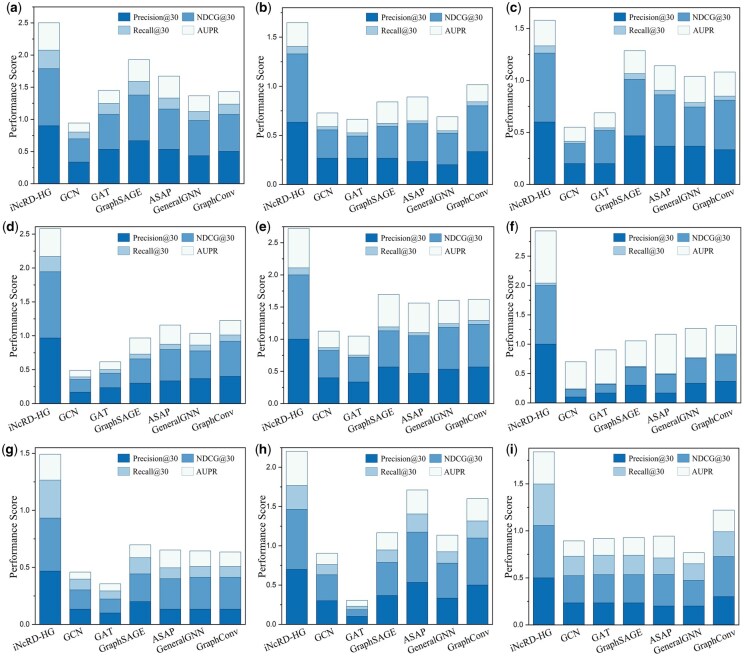 Figure 6
Figure 6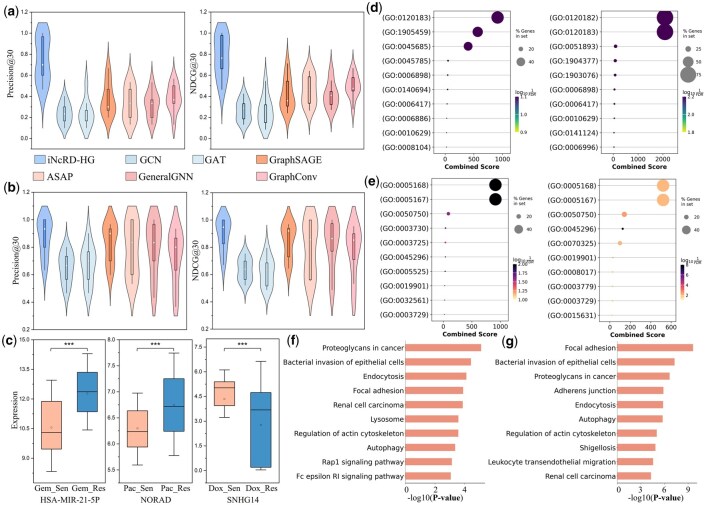 Figure 7
Figure 7| Subnetwork type | #Node 1 | #Node 2 | #Edge |
|---|---|---|---|
| lncRNA similarity | 1322 lncRNAs | 1322 lncRNAs | 8772 |
| miRNA similarity | 605 miRNAs | 605 miRNAs | 16232 |
| drug similarity | 216 drugs | 216 drugs | 852 |
| lncRNA-miRNA | 1322 lncRNAs | 605 miRNAs | 1841 |
| miRNA-drug resistance | 605 miRNAs | 200 drugs | 5394 |
| lncRNA-drug resistance | 1322 lncRNAs | 71 drugs | 2109 |
| Dataset | Method | AUC | AUPR |
|---|---|---|---|
|
| iNcRD-HG |
|
|
| w/o_LncMi | 0.9729 | 0.9242 | |
| w/o_attribute | 0.9699 | 0.9282 | |
| HG_SeqSim | 0.9718 | 0.9403 | |
|
| iNcRD-HG |
|
|
| w/o_ LncMi | 0.9518 | 0.9098 | |
| w/o_attribute | 0.9492 | 0.9062 | |
| HG_SeqSim | 0.9480 | 0.9084 |
| Method |
|
| ||
|---|---|---|---|---|
| AUC | AUPR | AUC | AUPR | |
| GAT | 0.9652 | 0.9273 | 0.8917 | 0.7219 |
| GraphSAGE | 0.9713 | 0.9202 | 0.9383 | 0.8718 |
| ASAP | 0.9525 | 0.8939 | 0.9160 | 0.8787 |
| GeneralGNN | 0.9615 | 0.9127 | 0.9226 | 0.8644 |
| GraphConv | 0.9469 | 0.9148 | 0.8989 | 0.8293 |
| GCMDR | 0.9634 | 0.9430 | 0.9123 | 0.8554 |
| DeepLDA | 0.9729 | 0.9335 | 0.9393 | 0.8850 |
| AGCLNDA | 0.6646 | 0.7643 | 0.9244 | 0.8871 |
| NDSGCL | 0.7723 | 0.7679 | 0.8008 | 0.6732 |
| iNcRD-HG |
|
|
|
|
| Drug | lncRNA | Evidence | miRNA | Evidence |
|---|---|---|---|---|
| Gemcitabine | 33436545 | HSA-MIR-21-5P | 16762633 | |
| HAO2-IT1 | 33436545 | HSA-MIR-34A-5P | 21347785 | |
| LINC01093 | 33436545 | HSA-MIR-200C-3P | 19654291 | |
| LINC01831 | 33436545 | HSA-MIR-181A-5P | 28560603 | |
| CPS1-IT1 | 33436545 | HSA-MIR-126-3P | 31514732 | |
| 33436545 | HSA-MIR-375-3P | 31514732 | ||
| 33436545 | HSA-MIR-203A-3P | 24040438 | ||
| LINC01595 | 33436545 | HSA-MIR-210-3P | 25117811 | |
| 33436545 | HSA-MIR-145-5P | 27765914 | ||
| 33436545 | HSA-MIR-100-5P | 23946872 | ||
| Paclitaxel | NEAT1 | 30782035 | HSA-MIR-93-5P | 30244336 |
| XIST | 12492109 | HSA-MIR-16-5P | 20460378 | |
| MALAT1 | 31837057 | HSA-MIR-15A-5P | 23826416 | |
| KCNQ1OT1 | 28600629 | HSA-MIR-26B-5P | 29125598 | |
| H19 | 29968942 | HSA-MIR-103A-3P | 30774764 | |
| OIP5-AS1 | 29219179 | HSA-MIR-101-3P | 24755562 | |
| SNHG16 | 31287002 | HSA-MIR-32-5P | 24220856 | |
| HOTAIR | 29680837 | HSA-MIR-301A-3P | 26323677 | |
| UCA1 | 29777711 | HSA-MIR-25-3P | 26323677 | |
| NORAD | unconfirmed | HSA-MIR-106B-5P | 20460378 | |
| Doxorubicin | NEAT1 | 30349370 | HSA-MIR-21-5P | 19412672 |
| XIST | 30907503 | HSA-MIR-34A-5P | 18645025 | |
| MALAT1 | 27524242 | HSA-MIR-200C-3P | 19412672 | |
| KCNQ1OT1 | 31837329 | HSA-MIR-181A-5P | 19412672 | |
| SNHG16 | unconfirmed | HSA-MIR-126-3P | 19237188 | |
| NORAD | 32563146 | HSA-MIR-146A-5P | 19412672 | |
| SNHG14 | unconfirmed | HSA-MIR-155-5P | 18971180 | |
| OIP5-AS1 | 30204936 | HSA-MIR-29A-3P | 18645025 | |
| FGD5-AS1 | unconfirmed | HSA-MIR-29B-3P | 18645025 | |
| H19 | 8674037 | HSA-MIR-451A | 19412672 |
- —National Natural Science Foundation of China10.13039/501100001809
- —Entrepreneurship Talent Project of Qin Chuang Yuan
- —Fundamental Research Funds for the Central Universities10.13039/501100012226
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCancer-related molecular mechanisms research · Machine Learning in Bioinformatics · MicroRNA in disease regulation
1. Introduction
Chemotherapeutic drugs serve as a cornerstone in the treatment of diseases, especially for malignant tumors. However, their therapeutic efficacy is frequently compromised by challenges such as tumor recurrence or progression post-treatment in most patients. A pivotal factor underlying therapeutic failure is the emergence of drug resistance, a multifactorial process orchestrated through diverse mechanisms like drug efflux pumps, target alterations, DNA repair enhancement, tumor microenvironment remodelling, and bypass alternative signalling pathway activation (Khan et al. 2024, Lin 2024, Patel et al. 2024, Yan et al. 2024, Qi et al. 2025, Qiao et al. 2025). This resistance phenotype stems from the crosstalk between molecular pathways, which collectively enable tumor cell survival under pharmacological pressure (Musyuni et al. 2022).
Non-coding RNAs (ncRNAs), as critical regulators of cellular processes, have been well-documented to participate in diverse physiological and pathological activities (Tang et al. 2018, Nemeth et al. 2024, Qiao et al. 2024). Recent studies have increasingly highlighted their fundamental involvement in mediating drug resistance (Chen et al. 2024, Wang et al. 2024). For examples, miR-23a-3p upregulation in an ETS1-dependent manner promotes sorafenib resistance in HCC, whereas its CRISPR-Cas9 knockout restores drug sensitivity (Lu et al. 2022). In multiple myeloma, H19 drives bortezomib resistance via the miR-29b-3p/MCL-1 axis (Pan et al. 2019). Identifying ncRNA-mediated drug resistance mechanism is critical for facilitating drug screening, developing tailored strategies against resistance, and discovering novel druggable targets within ncRNA-mediated regulatory networks.
The growing accumulation of biological data enables computational methods that exploit prior associations to identify ncRNA associated with drug resistance, offering guidance for experimental validation (Mahapatra et al. 2024, Chen et al. 2025, Lai et al. 2025). These methods fall into two main categories: pan-ncRNA predictors and models tailored to specific ncRNA types. Most pan-ncRNA predictors regard diverse ncRNAs as homogeneous nodes and leverage prior ncRNA–drug resistance associations within graph learning frameworks. Their main challenge is the highly sparse supervision signals of the prior bipartite network. GSLRDA addressed this by integrating dropout and random walk–based contrastive networks with lightweight GCN and self-supervised learning to enhance node representations (Zheng et al. 2022, Ren et al. 2025). NDSGCL further integrated local structural and global semantic neighbours for data augmentation (Hu et al. 2024), while AGCLNDA introduced adaptive graph contrastive learning by constructing enhanced and denoised bipartite graphs to improve prediction performance (Fan et al. 2025). Computational methods focusing on specific ncRNA types, primarily miRNAs and lncRNAs, have been developed, with research on miRNA–drug resistance garnering more attention. GCMDR integrated miRNA expression and functional similarity to represent miRNA attributes and used drug substructure fingerprints to characterize drugs, leveraging GCN to uncover potential associations from a heterogeneous miRNA–drug association network (Huang et al. 2020). SDNEMDA combined BiGRU and SDNE to integrate node attribute and topological features (Wang et al. 2024, Lei and Lei 2025), while DLST-MDA employed multi-scale CNN sequence encoder and GCN structure encoder to capture high-level miRNA and drug representations (Sheng et al. 2025). In contrast, research on lncRNA-drug resistance remains limited. DeepLDA employed the GIP strategy to construct lncRNA and drug similarity networks and utilized GCN and GAT to extract high-level representations, achieving promising predictive performance (Wei et al. 2021, Gao and Shang 2023, Yan et al. 2023, Ai et al. 2024).
Existing graph learning–based computational methods have advanced our understanding of the roles of ncRNAs in drug resistance mechanisms. Nevertheless, several challenges remain. (i) Although pan-ncRNA predictors design different contrastive learning frameworks to alleviate sparsity and noise in the original bipartite network, they still rely heavily on limited prior signals. Consequently, the biological semantic information embedded in the constructed contrastive networks is restricted and prone to semantic distortion. (ii) Whether pan-ncRNA predictors or models tailored to specific ncRNA types, most approaches focus exclusively on bipartite associations between individual ncRNAs and drugs. In reality, ncRNAs exert their functions via intricate molecular interactions that impact drug efficacy. Current methods overlook these synergistic ncRNA interactions, yielding networks devoid of molecular interaction context and unable to capture drug response-related molecular pathways. Overall, integrating biological contextual semantics to improve predictive reliability and interpretability remains a critical challenge (Liu et al. 2024).
To investigate the multifaceted roles of ncRNAs in drug resistance mechanisms, we propose a novel pan-ncRNA predictor, iNcRD-HG, for predicting ncRNA–drug resistance associations. iNcRD-HG enhances the biological semantics of prior sparse network by incorporating multi-source biological information from both node attributes and topological structures. Specifically, it integrates cooperative ncRNA interactions, intra-type molecular associations, specific expression profiles of ncRNAs across diverse cancer cell lines, and drug Simplified Molecular-Input Line-Entry System (SMILES) to construct a heterogeneous biological network. On this basis, we design a semantics-enhanced heterogeneous graph learning architecture that employs relation-type–specific modules with dynamic message passing to capture semantic dependencies between nodes, while incorporating an interpretability mechanism to infer biologically meaningful paths underlying predictions. Experimental results indicate that the constructed heterogeneous network effectively encodes rich biological semantics, enabling iNcRD-HG to achieve more reliable predictions of ncRNA–drug resistance associations.
2. Materials and methods
2.1 Datasets
The experimentally validated ncRNA–drug resistance associations were retrieved from ncRNADrug (Cao et al. 2024). To better encode the attributes of ncRNAs and drugs, ncRNA expression profiles were extracted from CCLE (Barretina et al. 2012, Ghandi et al. 2019), and drug SMILES strings were obtained from PubChem (Kim et al. 2025). Duplicate entries in ncRNADrug, along with ncRNAs and drugs lacking expression profiles or SMILES strings, were excluded. Following preprocessing, we obtained 1322 lncRNAs, 605 miRNAs, 216 drugs, 2109 lncRNA–drug resistance associations, and 5394 miRNA–drug resistance associations. The resulting ncRNA–drug resistance association datasets utilized for model training and evaluation can be represented as follows:
where and denote the known lncRNA-drug and miRNA-drug resistance associations, respectively. and represent negative pairs randomly generated from distinct extensive sets of unknown pairs, respectively, with their quantities being twice those of the corresponding positive associations. The dataset and can be further partitioned into training, validation, and testing sets in 0.6:0.2:0.2 ratio, as described below:
where training set is used to train model, validation set is employed for hyperparameter tuning, and testing set is reserved for final performance evaluation. No overlapping ncRNA–drug resistance pairs exist among the three subsets. Detailed statistical information for each sub-dataset is provided in Supplementary Table S1, available as supplementary data at Bioinformatics online. The datasets can be accessed and downloaded from https://zenodo.org/records/16919628.
2.2 The iNcRD-HG model architecture
iNcRD-HG is a graph-learning based framework to predict pan-ncRNA-drug resistance associations. As illustrated in Fig. 1, it comprises two main modules: heterogeneous network construction and context-enriched heterogeneous graph learning. The details of each module are described in the following sections.
The architecture of iNcRD-HG. (a) Heterogeneous network construction. The heterogeneous network is constructed based on ncRNA expression profiles, drug SMILES strings, validated ncRNA-drug resistance associations, and lncRNA-miRNA interactions. (b) Context-enriched heterogeneous graph learning. Node representations of ncRNAs and drugs are learned through the heterogeneous graph encoder. The edge decoder generates pairwise representations by concatenating ncRNA and drug features and predicts association scores using a multi-layer perceptron.
2.3 Heterogeneous network construction
To enrich the biological semantics of original ncRNA-drug resistance bipartite network, we performed attribute feature encoding for ncRNAs and drugs and constructed multi-relational subnetwork, thereby forming the heterogeneous association network shown in Fig. 1a.
To characterize ncRNAs within pathological contexts, we extracted expression profiles of miRNAs and lncRNAs across 938 cancer cell lines from CCLE (Barretina et al. 2012, Ghandi et al. 2019) and used them as initial node features for ncRNAs. For drugs, we encoded molecular structures using SMILES strings and derived their representations with ChemBERTa (Chithrananda et al. 2020), a transformer-based model pretrained on 77 million SMILES strings from PubChem (Kim et al. 2025) through masked language modeling and multi-task regression, enabling the capture of rich and generalizable chemical semantics.
Considering the relationships between different molecules, we constructed six multi-relational subnetworks, including lncRNA similarity network, miRNA similarity network, drug similarity network, lncRNA-miRNA interaction network, miRNA-drug resistance association network, and lncRNA-drug resistance association network. The ncRNA similarity networks were derived using Kendall’s tau coefficient based on the 938-dimensional expression profiles of miRNAs and lncRNAs. Drug similarity was calculated using the Tanimoto coefficient applied to drug SMILES strings. To ensure the high quality of the similarity networks, only edges with similarity scores greater than 0.5 were retained across all similarity networks. The lncRNA-miRNA interaction network was constructed by integrating experimentally validated interactions from RNAInter (Kang et al. 2022), NPInter (Zheng et al. 2023), and ENCORI (Li et al. 2014). The two ncRNA-drug resistance association networks were built using the curated prior associations in and , retrieved from the ncRNADrug (Cao et al. 2024). Table 1 presents the statistical information of each subnetwork comprising the overall heterogeneous network.
2.4 Context-enriched heterogeneous graph learning
2.4.1 Heterogeneous graph encoder
As heterogeneous graph learning excels at extracting rich semantics from complex biological networks (Liu et al. 2019, Yu et al. 2024, Zhang et al. 2024, Su et al. 2025), we developed a heterogeneous graph encoder based on relation-type-aware message passing modules for capturing multi-relational structure semantic of the constructed heterogeneous network. Different from standard Graph Convolutional Networks (GCN) layer (Kipf 2017, Li et al. 2021), General Convolution (GeneralConv) separately learns self-node and neighbour-specific transformation weights (You et al. 2020), enabling more flexible and expressive feature integration in noisy biological networks. Therefore, general convolution was used as the base operator for node representation learning. Formally, for each graph, the update rule is:
where denotes the representation of node at layer , is its neighbourhood, and are learnable weights for self and neighbour contributions, respectively, and denotes the LeakyReLU activation function.
There are six undirected edge types in the constructed heterogenous network, where represent lncRNA similarity, miRNA similarity, drug similarity, lncRNA-miRNA interaction, lncRNA-drug association, miRNA-drug association, respectively. To exploit the semantic diversity, we extended general convolution into relation-type-aware modules. For each edge type , a relation-type-aware message function is defined as:
where denotes neighbours of under relation , and and are relation-type-aware learnable parameters. Thus, for each node type, the updated embeddings are computed as:
where denotes the message update function along relation <x-y>, ⊕ denotes the aggregation operator, which can be instantiated using pooling functions such as max, mean, sum, or min. By employing relation-type-aware general convolution modules, heterogeneous graph encoder dynamically models both intra-type similarities and cross-type dependencies. This semantic augmentation enables deeper integration of molecular contexts and enhances the expressiveness of ncRNA and drug embeddings.
2.4.2 NcRNA-drug resistance association prediction
The edge decoder was designed to predict the ncRNA-drug resistance association scores. Through the heterogeneous graph encoder, we obtained high-level embeddings for lncRNAs, miRNAs, and drugs. Taking miRNA–drug resistance association prediction as an example, for a target miRNA–drug pair, a concatenation pooling layer was applied to capture the joint association features:
Subsequently, a multilayer perceptron (MLP) with three fully connected layers was employed to predict the association score:
where , and are three learnable parameter matrices, and is the miRNA-drug pair feature matrix. Concatenation pooling allows direct integration of miRNA and drug embeddings, preserving their distinct feature spaces, while the MLP captures complex nonlinear interactions between them, enabling more accurate modeling of association patterns. The binary cross-entropy is defined as the loss function for training iNcRD-HG:
where N is the number of training pairs, is the sigmoid activation. and denotes the predicted association score and ground truth label, respectively.
2.5 Performance evaluation
Two metrics including Area Under the Receiver Operating Characteristic Curve (AUC) and Area Under the Precision–Recall Curve (AUPR) are used to comprehensively evaluate the performance of different predictors (Guo et al. 2024, Huang et al. 2024, Zhu et al. 2024, Dao et al. 2025). In addition, because top-ranked predictions are typically more reliable and practically relevant, we also incorporate three top-k ranking metrics: Precision@k, NDCG@k, and Recall@k, which respectively evaluate the precision, normalized discounted cumulative gain (NDCG), and recall within the top-k predictions, providing a more fine-grained evaluation of predictive performance in practical settings. Detailed descriptions of these evaluation metrics are provided in the Supplementary Method, available as supplementary data at Bioinformatics online.
3. Results and discussion
3.1 Parameter sensitivity analysis for robustness evaluation
To evaluate the robustness of iNcRD-HG, parameter sensitivity analysis was performed focusing on four critical hyperparameters: the learning rate, the number of training epochs, the hidden channel dimension in heterogeneous graph learning, and the aggregation strategy for message passing across different edge types. This analysis was conducted using the validation dataset , with the lncRNA–drug resistance association prediction task as an example. The results are presented in Fig. 2, revealing the following observations: (1) The learning rate is found to exert relatively pronounced effect on training iNcRD-HG. A low learning rate results in slow convergence and susceptibility to local optima, whereas an excessively high rate causes unstable training and impedes convergence to a satisfactory global optimum. Thus, selecting an appropriate learning rate is essential for effective and stable training; (2) Regarding the number of epochs and hidden channel dimension, iNcRD-HG performs relatively stable within suitable ranges, underscoring its resilience to such variations; (3) For the aggregation strategies employed in message passing across multiple edge types, we observe that the max pooling strategy consistently yields inferior performance. This can likely be attributed to its tendency to diminish the contribution of relation-type-aware semantic information. By contrast, the mean, sum, and min aggregation strategies produce comparable results, indicating that their influence on the overall predictive performance is largely interchangeable. Collectively, these findings indicate that iNcRD-HG exhibits strong robustness to variations in its key hyperparameters. Based on a grid search, we subsequently determined the optimal configuration for the lncRNA–drug resistance association prediction task as follows: {learning rate = 0.0005, epochs = 100, hidden channel dimension = 64, aggregation strategy=’min’}. In addition, the same parameter search space was applied to miRNA–drug resistance association prediction task, with the corresponding results are presented in Supplementary Fig. S1, available as supplementary data at Bioinformatics online.
Parameter analysis of iNcRD-HG. AUC and AUPR obtained with different learning rates, training epochs, hidden dimensions, and aggregation strategies on SLDval.
3.2 Semantic enrichment boosts predictive capability
To evaluate the effectiveness of the semantic enrichment strategy, which combines attribute feature encoding for ncRNAs and drugs with the construction of multi-relational subnetworks to enhance the initially sparse prior association network, we conducted ablation experiments to compare iNcRD-HG with three baseline variants: w/o_LncMi, w/o_attribute and HG_SeqSim.
The w/o_LncMi baseline trains the heterogeneous graph predictor on a reduced network that excludes ncRNA nodes and edges unrelated to the target task. For instance, in the case of lncRNA–drug resistance association prediction task, the network includes only three subnetwroks: lncRNA similarity, drug similarity, and lncRNA–drug resistance association subnetworks. Similarly, for the miRNA–drug resistance association prediction task, only miRNA similarity, drug similarity, and miRNA–drug resistance association subnetworks are retained. The w/o_attribute baseline removes all task-specific attribute features from the nodes and instead uses one-hot encoded vectors as initial node representations, thereby omitting the semantic information introduced through attribute feature encoding. HG_SeqSim replaces the original expression-based ncRNA similarity networks in the heterogeneous graph with sequence-based similarity networks. Specially, lncRNA and miRNA sequences were obtained from RNAcentral (Consortium 2021). These sequences were embedded using Word2Vec (Church 2017), and ncRNA similarity was also computed via Kendall’s tau coefficient.
The comparative results are presented in Table 2, from which we can see the followings: (1) Across both datasets, iNcRD-HG consistently outperforms the three baseline models, achieving the highest AUC and AUPR scores. These results highlight the importance of incorporating semantic information derived from related ncRNA subnetworks and attribute features in enhancing predictive performance; (2) Integrating cross-ncRNA interactions leads to a more pronounced performance improvement in lncRNA–drug resistance prediction than in miRNA–drug resistance prediction. Unlike miRNAs, which act primarily via direct post-transcriptional regulation, lncRNAs often function by competitively binding to miRNAs. Incorporating miRNA–lncRNA regulatory relationships therefore provides additional context that helps iNcRD-HG capture the indirect but important roles of lncRNAs in drug resistance; (3) Using sequence-based ncRNA similarity networks (HG_SeqSim) leads to slightly lower performance than iNcRD-HG, suggesting that expression profiles across cancer cell lines provide additional pathological context beyond sequence information for predicting ncRNA–drug resistance associations.
3.3 Feature analysis of heterogeneous graph representations
To further investigate the discriminative capability of ncRNA–drug resistance pair representations learned by iNcRD-HG, we performed visualization analyses using t-SNE. Three different pair representation strategies were compared. The first strategy, attribute concatenation, involves directly concatenating the attribute features of ncRNA and drug nodes. The second strategy, structural concatenation, integrates similarity vectors derived from ncRNA and drug similarity networks as the pair representation. The third strategy employs the pair representations extracted by iNcRD-HG through its heterogeneous graph learning framework, which jointly encodes node attribute features and topological dependencies across multi-relational molecular networks. As shown in Fig. 3, we visualized the pair representations produced by these three strategies on the testing datasets and . The results clearly demonstrate that attribute concatenation and structural concatenation show less separable clusters, whereas the representations learned by iNcRD-HG exhibit superior label separability. This improvement can be attributed to iNcRD-HG ability to integrate intrinsic semantic information from ncRNA and drug attributes with the complex topological semantics of molecular interactions, further enhanced by the supervision provided by association signals during representation learning.
T-SNE visualization of different pair representations. (a, d) Features derived from attribute concatenation. (b, e) Features obtained through structural concatenation. (c, f) Features learned by iNcRD-HG.
3.4 Interpretable predictions of ncRNA–drug resistance associations
To elucidate the inference pathways of iNcRD-HG in predicting ncRNA–drug resistance associations and enhance its biological interpretability for guiding experimental validation, we introduced Captum Explainer (Kokhlikyan et al. 2020) to compute attribution scores by back-propagating through iNcRD-HG model. We applied CaptumExplainer to the testing datasets and , and evaluated the importance scores of different nodes and edges within the constructed heterogeneous network. Figure 4a summarizes the results and reveals several key findings: (1) Among node types, miRNAs exhibit the highest contribution in both tasks, particularly in miRNA–drug resistance association prediction (64.6%), while lncRNAs contribute more prominently in lncRNA–drug resistance association prediction (36.4%), reflecting the task-specific relevance of different ncRNA types; (2) All edge types contribute to the final prediction, with lncRNA–drug and miRNA–drug edges showing the greatest impact, underscoring the importance of direct ncRNA–drug resistance associations; (3) Notably, lncRNA–miRNA interactions also play a significant role in both tasks (24.3% and 25.7%, respectively), aligning with the known bio logical mechanism of ncRNA cross-talk.
Interpretability analysis of iNcRD-HG predictions. (a) Contribution of different node and edge types in heterogeneous network to iNcRD-HG prediction. (b) Edge-contribution subnetwork for experimentally validated AFAP1-AS1 associated with CISPLATIN resistance. (c) Edge-contribution subnetwork for experimentally validated HSA-MIR-372-3P associated with TAXANE resistance.
Two experimentally validated associations <AFAP1-AS1, CISPLATIN> and <HSA-MIR-372-3P, TAXANE>, were analysed using CaptumExplainer to identify key molecular components and interactions driving inference, with Fig. 4b and c showing their top-300 edge-contribution subnetworks. For <AFAP1-AS1, CISPLATIN>, iNcRD-HG highlights meta-paths such as <AFAP1-AS1, FLUOROURACIL, HSA-LET-7A-5P, CISPLATIN>, implying that AFAP1-AS1 may exert cisplatin resistance through interactions with shared chemoresistance-associated regulators like let-7a or chemotherapeutic cross-talk pathways. While AFAP1-AS1 has been experimentally shown to promote cisplatin resistance via sponging miR-139-5p and upregulating RRM2 (Huang et al. 2019), the inference of let-7a suggests additional or parallel regulatory axes, as let-7a is known to modulate apoptosis-related genes such as BCL2 and HMGA2, potentially converging on similar resistance mechanisms. For <HSA-MIR-372-3P, TAXANE>, several high-impact meta-paths are identified, such as <HSA-MIR-372-3P, SNHG16, HSA-LET-7C-5P, TAXANE> and <HSA-MIR-372-3P, NORAD, HSA-MIR-20A-5P, TAXANE>, suggesting that miR-372-3p may influence taxane resistance through regulatory cascades involving lncRNAs and shared miRNA hubs such as let-7 and miR-20a families. These findings are biologically plausible, as miR-372-3p was found dysregulated in taxane-resistant tumors (Chen et al. 2020), SNHG16 has been linked to chemoresistance through modulation of cell cycle regulators, and NORAD contributes to genomic stability by sequestering PUMILIO proteins (Dong et al. 2019, Elguindy and Mendell 2021), which may indirectly affect drug response pathways. Two additional validated associations <XIST, OXALIPLATIN> and <HSA-MIR-199B-5P, SORAFENIB> were analysed, and their edge-contribution subnetworks are shown in Supplementary Fig. S2, available as supplementary data at Bioinformatics online.
Overall, these results highlight that iNcRD-HG effectively integrates node-level semantic attributes with molecular interaction edges to capture biologically meaningful pathways, while meta-path interpretation elucidates plausible mechanisms of ncRNA–drug resistance and uncovers novel intermediate nodes that may constitute unexplored regulatory axes warranting experimental validation.
3.5 Performance comparison with related methods
To evaluate the predictive performance of iNcRD-HG for ncRNA–drug resistance associations, we compared it against nine competing methods. Among these, five are GNN baselines, including GAT (Veličković et al. 2018), GraphSAGE (Hamilton et al. 2017), ASAP (Ranjan et al. 2020), GeneralGNN (You et al. 2020), and GraphConv (Morris et al. 2019). These GNN-based models utilized the node attribute features extracted in this study and were trained on a biological network comprising ncRNA–drug resistance associations, ncRNA similarity and drug similarity associations, derived by removing non-target ncRNA nodes and their associated edges from our heterogeneous network. The other four competing methods are existing ncRNA–drug association prediction approaches: GCMDR (Huang et al. 2020), DeepLDA (Gao and Shang 2023), AGCLNDA (Fan et al. 2025), and NDSGCL (Hu et al. 2024). GCMDR and DeepLDA leverage ncRNA and drug node attribute features and employ GCN and DNN architectures to extract high-level representations, whereas AGCLNDA and NDSGCL rely on graph contrastive learning to capture topological representations without explicit node attributes. Table 3 summarizes the performance comparison on two testing datasets and . The results reveal the key observations: iNcRD-HG consistently outperforms all baselines across both datasets, achieving the highest AUC and AUPR scores, underscoring its ability to integrate attribute information with heterogeneous topological semantics for robust ncRNA–drug resistance association prediction. In contrast, graph contrastive learning methods exhibit unstable performance, particularly in , potentially linked to their reliance on topology, making them more vulnerable to noise and reducing generalization. Attribute-based methods and standard GNN models perform competitively but still lag behind.
3.6 Performance under unseen ncRNA or drug conditions
To assess model generalization, we conducted leave-one-ncRNA-out and leave-one-drug-out experiments, and compared iNcRD-HG with six GNN-based baseline methods, including GCN, GAT, GraphSAGE, ASAP, GeneralGNN, and GraphConv, under unseen ncRNA or drug conditions.
For the unseen ncRNA condition, both leave-one-lncRNA-out and leave-one-miRNA-out experiments were performed. LncRNAs and miRNAs with more than 10 known drug resistance associations in the datasets and were selected for evaluation. The performance distributions across four evaluation metrics obtained by different methods are shown in Fig. 5a and b, respectively. Under the unseen lncRNA setting, iNcRD-HG consistently outperforms all six baseline models across all evaluation metrics. Under the unseen miRNA setting, iNcRD-HG achieves higher AUPR values and exhibits ranking performance comparable to GraphConv, underscoring its ability to prioritize true associations and comprehensively identify potential ncRNA–drug resistance relationships.
Performance of different methods under unseen ncRNA conditions. (a) Performance distributions for the leave-one-lncRNA-out experiment. (b) Performance distributions for the leave-one-miRNA-out experiment.
For leave-one-drug-out experiment, nine chemotherapeutic drugs were selected for evaluation, as each exhibits more than 30 known ncRNA resistance associations in the datasets and . The drug-specific results are analyzed and illustrated in Fig. 6, from which we can see that iNcRD-HG achieves the best performance across all four metrics compared to six GNN-based baseline methods, with particularly notable improvements in Precision@30 and NDCG@30. This demonstrates iNcRD-HG can accurately prioritize true ncRNA-drug resistance associations and achieve higher ranking quality. From an overall perspective, all methods show generally higher Precision@30 and NDCG@30 compared to Recall@30. This phenomenon can be attributed to the limited number of known ncRNA-drugs associations: with relatively few true positives per drug and a fixed k value of 30, ranking precision and quality are more readily reflected, whereas recall are inherently constrained by the small pool of true associations. Comparative results of miRNA-drug prediction performance across the nine unseen drugs are provided in Supplementary Fig. S3, available as supplementary data at Bioinformatics online. The performance distributions of lncRNA-drug and miRNA-drug resistance association prediction are shown in Fig. 7a and b, with detailed mean metrics provided in Supplementary Tables S2 and S3, available as supplementary data at Bioinformatics online. iNcRD-HG outperforms all baselines in both tasks, achieving superior Precision@30 and NDCG@30, underscoring its ability to rank true associations at the top positions. These results demonstrate that iNcRD-HG excels not only in standard prediction scenarios but also maintains robustness under different cold-start conditions.
Performance of different methods in leave-one-drug-out experiments on SLD. (a–i) Evaluation metrics under unseen drug scenarios, with Doxorubicin, Oxaliplatin, Fluorouracil, Temozolomide, Cisplatin, Gemcitabine, Gefitinib, Paclitaxel, and Sorafenib each sequentially treated as the simulated novel drug.
Case study analysis of iNcRD-HG under simulated novel drug scenarios. (a and b) Leave-one-drug-out performance metrics for lncRNA- (a) and miRNA- (b) drug resistance association prediction tasks across different methods. (c) Expression levels of three predicted ncRNAs in drug-sensitive and drug-resistant cells for Gemcitabine (Gem), Paclitaxel (Pac), and Doxorubicin (Dox), where ‘_Sen’ and ‘_Res’ indicate drug-sensitive and drug-resistant cells, respectively. (d) GO Biological Process (BP) enrichments of ncRNAs associated with Gemcitabine and Paclitaxel resistance identified by iNcRD-HG. (e) GO Molecular Function (MF) enrichments of ncRNAs associated with Gemcitabine and Paclitaxel resistance identified by iNcRD-HG. (f and g) KEGG pathway enrichments of ncRNAs associated with Gemcitabine and Paclitaxel resistance identified by iNcRD-HG.
3.7 Case study
To further illustrate the predictive capability of iNcRD-HG under unseen drug conditions, we conducted case studies on three representative drugs including Gemcitabine, Paclitaxel, and Doxorubicin. Table 4 lists the top 10 lncRNAs and miRNAs predicted by iNcRD-HG as resistance-associated candidates for each drug. Of the top-ranked candidates, 26 lncRNAs and 30 miRNAs are supported by experimental evidence reported in the literature. For example, knockdown of NEAT1 reverses paclitaxel resistance in non-small cell lung cancer (NSCLC) cell lines (Li et al. 2019), inhibition of miR-21 increases sensitivity to gemcitabine (Meng et al. 2006), and NORAD promotes proliferation and metastasis while conferring doxorubicin resistance and inhibiting apoptosis and autophagy in neuroblastoma cells (Wang et al. 2020). Certain ncRNAs, including MALAT1, XIST, and HSA-MIR-34A-5P, appear recurrently across different drugs, suggesting their potential roles as shared regulators of multidrug resistance. Although several predicted ncRNAs lack direct experimental validation, NORAD and SNHG14 showed significant differential expression in paclitaxel- and doxorubicin-resistant versus sensitive cells, respectively (Fig. 7c). Together, these findings highlight that iNcRD-HG can not only recover known drug resistance-associated ncRNAs but also identify promising candidates with potential clinical relevance.
Potential biological mechanisms by which ncRNAs contribute to drug resistance were investigated through functional enrichment analysis of the top 100 candidates predicted by iNcRD-HG. As shown in Fig. 7d, ncRNAs associated with gemcitabine resistance are significantly enriched in GO Biological Process (BP) terms such as focal adhesion disassembly, apoptosis regulation, receptor-mediated endocytosis, and translational control, indicating that alterations in adhesion dynamics, intracellular transport, and gene expression regulation are central to gemcitabine response. In contrast, paclitaxel-related ncRNAs are enriched in processes involving focal adhesion assembly/disassembly, protein localization, intracellular signalling, and biosynthetic regulation, highlighting a stronger involvement of signalling modulation and membrane protein trafficking in paclitaxel resistance. Molecular function analysis (Fig. 7e) indicates common enrichment in cadherin binding, protein kinase binding, and mRNA binding, but gemcitabine resistance uniquely involved GTP and double-stranded RNA binding, while paclitaxel resistance is characterized by microtubule, tubulin, and actin binding, reflecting its mechanism as a microtubule-stabilizing drug. KEGG pathway analysis (Fig. 7f–g) indicates overlapping involvement of focal adhesion, endocytosis, autophagy, and actin cytoskeleton regulation, alongside drug-specific pathways such as Rap1 and Fc epsilon RI signalling for gemcitabine, and adherens junction and leukocyte transendothelial migration for paclitaxel. These results suggest that ncRNAs contribute to chemoresistance via shared pathways regulating adhesion, endocytosis, and cytoskeletal remodelling, while also engaging distinct biological processes aligned with each drug pharmacological action.
4. Conclusion
Deciphering how non-coding RNAs contribute to drug resistance is pivotal for revealing cellular response mechanisms and advancing targeted cancer therapies. In this work, we propose iNcRD-HG, a heterogeneous graph learning framework that integrates multi-source biological information, including molecular interactions, ncRNA expression patterns, and drug structural features, to construct a biologically enriched network. By leveraging relation-type-aware message passing and an interpretability module, iNcRD-HG captures intricate contextual relationships and uncovers cooperative ncRNA effects influencing drug resistance. Experimental results demonstrate iNcRD-HG achieves superior predictive performance compared with existing methods, effectively uncovering synergistic ncRNA interactions and biologically meaningful pathways associated with drug resistance.
Although iNcRD-HG demonstrates promising results, several avenues for improvement remain: (i) Incorporating multiple ncRNA modalities, including sequence and expression features, along with the validated circRNA–drug resistance associations, can enhance the representation of ncRNA and enable a more comprehensive exploration of ncRNA-mediated regulatory mechanisms. (ii) A sophisticated multi-task learning module can be introduced to extend iNcRD-HG, as naive multi-task designs may cause task interference. Potential strategies include cross-task consistency regularization leveraging known cross-ncRNA interactions, task-aware contrastive objectives, and adaptive loss reweighting to better preserve task-specific biological signals while regulating cross-task influence. (iii) Since iNcRD-HG is built on aggregated public databases, it currently provides coarse-grained drug resistance associations. Tumor-type-specific or single-cell information can be further integrated to improve tumor–specific interpretation.
Supplementary Material
btag029_Supplementary_Data
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Ai C , Yang H, Liu X et al MT Mol-GPT: de novo multi-target molecular generation with transformer-based generative adversarial imitation learning. P Lo S Comput Biol 2024;20:e 1012229.38924082 10.1371/journal.pcbi.1012229 PMC 11233020 · doi ↗ · pubmed ↗
- 2Barretina J , Caponigro G, Stransky N et al The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012;483:603–7.22460905 10.1038/nature 11003 PMC 3320027 · doi ↗ · pubmed ↗
- 3Cao X , Zhou X, Hou F et al nc RNA Drug: a database for validated and predicted nc RN As associated with drug resistance and targeted by drugs. Nucleic Acids Res 2024;52:D 1393–D 1399.37953323 10.1093/nar/gkad 1042 PMC 10767907 · doi ↗ · pubmed ↗
- 4Chen D , Bao C, Zhao F et al Exploring specific mi RNA-m RNA axes with relationship to Taxanes-Resistance in breast cancer. Front Oncol 2020;10:1397.32974144 10.3389/fonc.2020.01397 PMC 7473300 · doi ↗ · pubmed ↗
- 5Chen Y , Wang J, Wang C et al Auto Edge-CCP: a novel approach for predicting Cancer-Associated Circ RN As and drugs based on automated edge embedding. P Lo S Comput Biol 2024;20:e 1011851.38289973 10.1371/journal.pcbi.1011851 PMC 10857569 · doi ↗ · pubmed ↗
- 6Chen Y , Wang Z, Wang J et al Self-supervised learning in drug discovery. Sci China Inf Sci 2025;68:170103.
- 7Chithrananda S , Grand G, Ramsundar B. Chem BER Ta: large-scale self-supervised pretraining for molecular property prediction. ar Xiv 2010.09885, 2020, preprint: not peer reviewed.
- 8Church KW. Word 2Vec. Nat Lang Eng 2017;23:155–62.