The whole genome sequencing offers insights into the susceptibility to the foot-and-mouth disease virus carrier state
Zhihui Zhang, Zhidong Teng, Shuanghui Yin, Suyu Mu, Sumin Wei, Yaozhong Ding, Yun Zhang, Shuang Wang, Yijing Li, Shiqi Sun, Huichen Guo

TL;DR
This study identifies genetic variants in cattle that may influence susceptibility to persistent foot-and-mouth disease virus infection.
Contribution
The study provides a catalog of genetic variants linked to viral persistence in cattle through whole-genome sequencing.
Findings
24 shared carrier-specific genetic variants were identified across cattle genomes.
31 carrier-specific variants were found in cattle–yak haplotypes, affecting immune-related genes.
These variants may influence immune regulation and contribute to viral persistence.
Abstract
Foot-and-mouth disease virus (FMDV) establishes persistent infection in more than 50% of infected ruminants, irrespective of vaccination status, implicating potential contributions of host genetic variations to viral persistence. In this study, we conducted whole-genome resequencing of a cohort of 22 cattle, comprising 7 carriers and 15 noncarriers. Clean reads were mapped to the cattle (Bos taurus) reference genome (ARS-USD1.2) and cattle–yak (Bos taurus × Bos grunniens) haplotype assemblies. We identified 24 shared carrier-specific variants across genomes and 31 carrier-specific variants restricted to the cattle–yak haplotypes. These carrier‑specific variants were primarily located in genes involved in olfactory perception, cell development and morphological maintenance, transcriptional and translational regulation, signal transduction, metabolic homeostasis, stress resistance, and…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
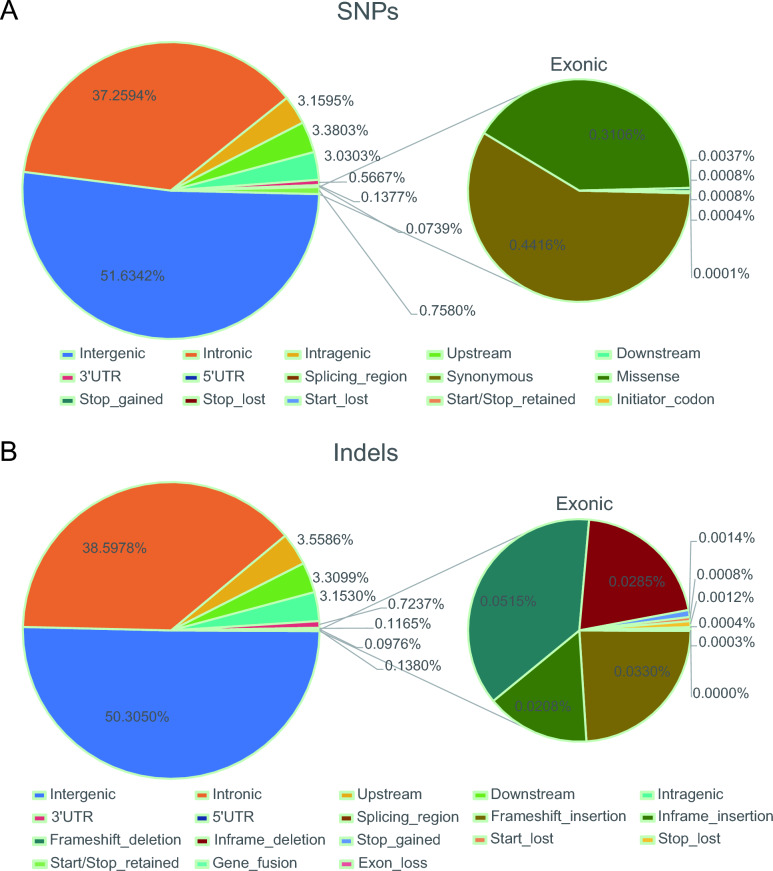 Figure 1
Figure 1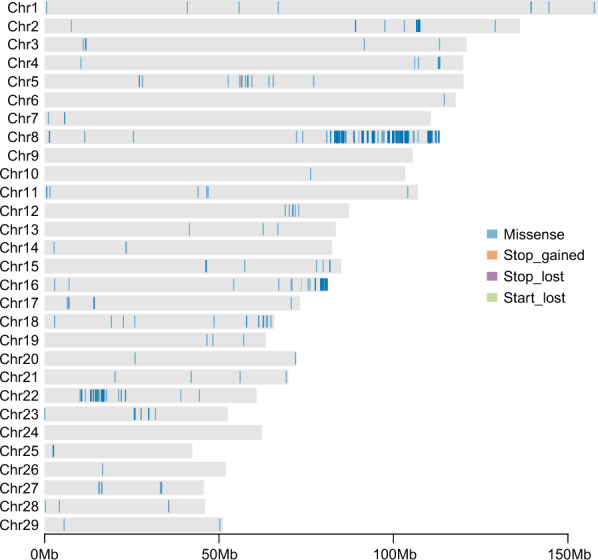 Figure 2
Figure 2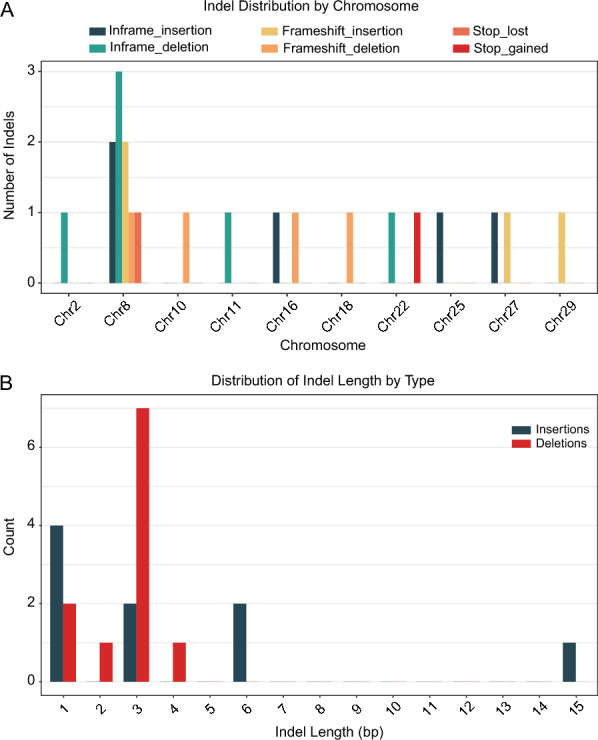 Figure 3
Figure 3 Figure 4
Figure 4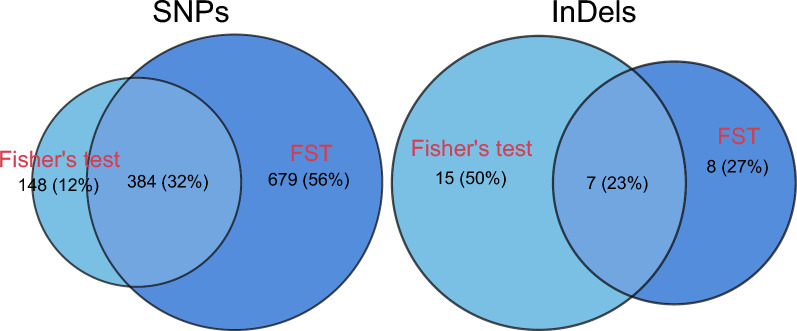 Figure 5
Figure 5- —National Key R&D Program of China
- —Technology innovation guidance program of Gansu Province
- —thereby acting
- —Key R&D Program of Ningxia province
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAnimal Disease Management and Epidemiology · Viral Infections and Immunology Research · Animal Virus Infections Studies
Introduction
Foot-and-mouth disease (FMD) is a highly contagious and economically devastating viral disease of livestock, caused by infection with FMD virus (FMDV), an Aphthovirus within the Picornaviridae family. The disease affects more than 70 species of wild and domestic cloven-hoofed animals, including major livestock species such as cattle, pigs, sheep, and goats, as well as buffalo and wild boar [1]. In FMD-endemic regions, substantial annual losses arise from vaccination costs, enforcement of animal health measures, and trade restrictions [2, 3]. Conversely, FMD-free countries invest heavily in preparedness and surveillance, as any outbreak could severely disrupt livestock production and animal product trade [4]. FMD presents exceptional challenges to control owing to its minimum infectious dose, rapid viral replication and broad host range, which is further compounded by subclinical persistent infection in ruminants following viral exposure [5]. The persistent FMDV infection, also known as the FMDV carrier status, is defined as the recovery of the virus from oropharyngeal fluid (OPF) more than 28 days post-infection by the World Organization for Animal Health (WOAH) [6].
Clinical investigations have documented an incidence exceeding 50% for FMDV persistence, even among herds fully protected by vaccination [7–9]. The duration of the carrier state varies among animal species, lasting up to 9 months in sheep, 3.5 years in cattle, and 5 years in African buffalo [10]. The anatomical location of viral persistence also varies among animal species. In cattle, FMDV is primarily confined to the nasopharynx [7]. In sheep, it persists within the epithelial crypts of the oropharyngeal and laryngeal tonsils [11]. In African buffalo, viral persistence is observed in the pharyngeal and palatine tonsils, as well as the nasopharyngeal mucosa [12]. Critically, experimental data demonstrate that transmission from carriers occurs [13–16]. Although the individual transmission probability appears minimal [17], the high prevalence of FMDV infection and frequent cross-border animal movements amplify the number of carriers and heighten population-level epidemiological concerns. Moreover, heterologous FMDV superinfection in carrier cattle frequently drives viral recombination in the upper respiratory tract, generating dominant interserotype recombinants [18, 19]. The shedding of such recombinant viruses may lead to subclinical infections without overt clinical manifestations. These epidemiological and virological factors make carrier animals a significant barrier to FMD control, eradication, and attainment of WOAH FMD-free status.
The viral genetic determinants of FMDV persistence have been extensively investigated, yet most studies have failed to identify specific genomic features correlated with the carrier state [20–23]. Although particular type O strains exhibit specific amino acid substitutions, such as VP1 Q172R (ME-SA/PanAsia) [24], VP2 Y80H (UKG34/2001) [25], VP2 Y79H and VP3 A75T (FRA/1/2001) [26], these associations lack further corroboration. Consistent with SAT1/KNP/196/91 strain in buffaloes [20], persistent isolates of O/FRA/1/2001 strain show no evidence of antigenic escape from neutralizing antibodies [26]. Litz et al. reported that leaderless O/FRA/1/2001 failed to cause clinical disease or persistence in cattle [27], though it is unclear whether similar effects occur across other strains or serotypes. While SAT1/KNP/196/91 strain exhibited greater prevalence and longer persistence than SAT2/KNP/19/89 and SAT3/KNP/1/08 strains in coinfected African buffalo [12], no competitive advantage was observed between O1 Manisa and A24 Cruzeiro strains in coinfected cattle [18, 28]. Collectively, these findings suggest that while viral determinants may contribute to persistence, host factors play a decisive role, as in vivo FMDV replication in cattle triggers strong antiviral responses [9, 29–31].
In carrier cattle, antiviral responses in microdissected FMDV-positive nasopharyngeal epithelium are locally suppressed, without obvious histopathological lesions [8], suggesting a localized virus–host equilibrium. Viral clearance is primarily driven by adaptive immunity, with FMDV infection and vaccination predominantly inducing robust antibody-mediated responses. However, systemic antibody titers did not differ between carriers and noncarriers [9, 29]. Although carriers exhibit stronger FMDV-specific IgA responses in nasal and oral secretions [32–34], they remain unable to eliminate the virus sequestered within host cells. Transcriptome analyses of microdissected nasopharyngeal epithelium have linked the carrier state to the impaired cell-mediated immunity [35, 36], as noncarriers exhibit higher T cell infiltration within nasopharyngeal mucosa [35], and cellular immune responsiveness of peripheral blood mononuclear cells inversely correlates with viral persistence [37]. Whether host-intrinsic factors govern clearance versus persistence remains unclear.
Interindividual genetic variations can influence both susceptibility to and clinical progression of viral infection. Polymorphisms in innate immune-related genes (OAS1 [38], IFNAR1 [39], MxA [40], and IRTK4 [41]) and viral receptor genes (SCARB2, PSGL1, and ANXA2) [42] have been linked to the severity of enterovirus 71-induced hand-foot-and-mouth disease. Moreover, loss-of-function mutations of IFIH1 increase vulnerability to respiratory RNA viruses, including respiratory syncytial virus and rhinoviruses [43]. Regarding FMDV, polymorphisms in the BoLA-DRB3 gene [44, 45] or in integrin genes [46, 47] potentially influence disease susceptibility. Five BoLA-DRB3 genotypes have been reported in Egyptian buffalo populations, with the AA genotype associated with FMD resistance and the AC genotype with greater FMD susceptibility [48]. Genome-wide association studies (GWAS) have demonstrated that resistance to FMDV in Holstein cattle is associated with enhanced innate immune responses [49]. Compared with Holstein–Friesian (Bos taurus) cattle, two Indian breeds–Malnad Gidda and Hallikar (Bos indicus)–showed a significant upregulation of genes involved in mitochondrial activation and innate antiviral immune pathways during acute infection, leading to reduced viral loads and earlier induction of neutralizing antibody and interferon-gamma (IFN-γ) responses [50]. Therefore, a comprehensive understanding of host genetic determinants underlying persistent FMDV infection informs virus–host coevolution studies and supports host-targeted strategies, including disease-resistance breeding and personalized modulation.
To this end, we conducted whole-genome sequencing (WGS) to identify differential genetic variations between vaccinated carriers and noncarriers. We propose a set of candidate variants potentially implicated in the establishment of persistent FMDV infection. Our findings provide novel insights into the genetic architecture underlying viral persistence and offer avenues for improved prevention and control strategies.
Materials and methods
Samples and sequencing
Peripheral blood was collected from 22 vaccinated cattle, comprising 7 FMDV carriers and 15 noncarriers. The detailed demographic and clinical characteristics of the enrolled subjects are provided in Additional file 1. Genomic DNA was extracted via the DNeasy Blood and Tissue Kit (Qiagen) following the manufacturer’s protocol. The concentration and quality of the extracted genomic DNA were assessed via a NanoDrop 2000 Spectrophotometer (Thermo Fisher Scientific). Sequencing libraries were prepared using the NEBNext DNA Library Prep Reagent Set (BioLabs, no. E6000). High-throughput sequencing was conducted on an Illumina NovaSeq 6000 platform (Illumina, CA, USA), generating 150-bp paired-end reads for downstream analyses.
Reads alignment
To minimize artificial bias in sequencing reads, raw reads were filtered out according to the following criteria: (1) the content of unidentified nucleotides was greater than 10%; (2) reads with > 10 nucleotides aligned to the adapter, allowing ≤ 10% mismatches; and (3) average base-quality less than 20 (Phred-like score). Finally, the N Tb (Q30 = 90.0%) values of the high-quality sequences were obtained for subsequent analyses. The clean paired-end reads were then aligned to the cattle (Bos taurus) reference genome (ARS-USD1.2) via the maximum entropy method (MEM) algorithm of Burrows-Wheeler Aligner (BWA) software (v.0.7.8) with the optimized parameters (‘men -t 4 -k 32 -M’). The resulting alignment files in SAM format were processed via SAMtools (v.1.3) to convert, index, and sort the data, ultimately generating BAM files. If multiple read pairs had identical external coordinates, only the pair with the highest mapping quality was retained. Potential PCR duplicates were removed via Picard (v1.9.4) to improve alignment results.
To assess potential sex-related effects on the analysis, we retrieved the sex-determining region Y (SRY) gene sequences of cattle and yak from the Ensembl website. Clean reads were aligned to the SRY reference sequences. Substantial read coverage across multiple Y-specific regions was considered indicative of male individuals.
SNPs/InDels calling and annotation
After alignment, single nucleotide polymorphisms (SNPs) and short insertions and deletions (InDels, 1–49 bp) for all individuals were called simultaneously using the Genome Analysis Toolkit (GATK) HaplotypeCaller (release 4.0). SNPs were filtered using the following criteria: variant quality by depth (QD) < 2.0; Phred-scaled p-value using Fisher’s exact test to detect strand bias (FS) > 60.0; root mean square of the mapping quality score (MQ) < 40.0; strand odds ratio of 2 × 2 contingency table to detect strand bias (SOR) > 3.0; mapping qualities of Ref reads (MQRankSum) < −12.5; and ranked-sum test for the distance of alleles from the end of the reads (ReadPosRankSum) < −8.0. We then filtered out allosomal SNPs and SNPs with a missing genotype rate > 0.1 and a minor allele frequency (MAF) < 0.05.
InDels were filtered using the following criteria: QD < 2.0; FS > 200.0; MQ < 40.0; SOR > 10.0; MQRankSum < −12.5; and ReadPosRankSum < −8.0. The autosomal InDels with a MAF ≥ 0.05 and a missing genotype rate of 0 were subsequently retained for further analyses.
Identification of differential variants between carriers and noncarriers
Among all autosomal variants from 22 cattle retained after quality filtering, we selected missense variants within splice donor sites and exonic regions to identify differential variants that were polymorphic between carriers and noncarriers via Fisher’s exact test. Variants exhibiting extremely statistical significance (p-value < 0.01) were retained for subsequent downstream analyses.
Allelic frequency differentiation between carriers and noncarriers
We performed fixation index (FST) analysis using VCFtools (0.1.16) [51] to quantify allele frequency differentiation between carrier and noncarrier groups across autosomes, focusing on missense variants located within splice donor sites and exonic regions. The variants with top 1% FST values were considered to be of strong selective pressure or genetic divergence between the two groups and were subjected to further analyses.
Determination of candidate regions or genes
The overlapping genomic regions, with both a p-value < 0.01 determined by Fisher’s exact test and a top 1% FST value, were subjected to gene annotation. Among overlapping regions, genes harboring variants exclusively observed in the carrier group were identified as candidate genes that differentiate carriers and noncarriers.
GWAS
To identify significantly associated SNPs with the carrier state, the R package rMVP (version 1.4.5) was applied for case–control GWAS. Principal component analysis (PCA) was performed to estimate and adjust population stratification. Kinship matrices were generated to calculate the pairwise kinship coefficients required to correct for relatedness among individuals using the identity-by-state (IBS) method. GWAS analyses were conducted using generalized linear model (GLM), mixed linear model (MLM), and FarmCPU model simultaneously, with a threshold of 5 × 10^−8^, incorporating both PCA results and the kinship matrix to adjust for population structure and relatedness. Manhattan and Quantile–quantile plots were generated to visualize GWAS results and assess the overall fit of the model, which confirmed proper control of inflation factors typically affected by population stratification and kinship. Significant associations were identified on the basis of a genome-wide significance threshold corrected for multiple testing.
Cattle–yak haplotype genome mapping
The cattle–yak (Bos taurus × Bos grunniens) haplotype 1 and haplotype 2 assemblies were generously provided by Professor Minghui Kang and Jianquan Liu, which remain unpublished to date. The clean reads were aligned to the cattle–yak (Bos taurus × Bos grunniens) haplotype 1 and 2 genomes separately. The missense variants within exonic regions and splice donor sites were selected to identify differential variants between carriers and noncarriers using Fisher’s exact test. The identified candidate variants based on the cattle (Bos taurus) reference genome (ARS-USD1.2) were mapped to these two haplotype genomes to assess potential artifacts arising from hybrid recombination.
Results
Genomic landscape of SNPs and InDels
A total of approximately 4.3 billion paired-end clean reads were generated from WGS of 22 cattle samples, with an average coverage depth of ~10× on the Illumina sequencing platform. These reads were aligned to the cattle (Bos taurus) reference genome (ARS-UCD1.2), attaining a high average genome coverage of 97.92% (Additional file 2). After quality filtering, we identified 32 883 797 autosomal SNPs with missing genotype rates < 0.1 and MAFs ≥ 0.05 (Additional file 3), and 2,834,320 InDels with missing genotype rates = 0 and MAFs ≥ 0.05 (Additional file 4). These high-quality variants were subjected to subsequent analyses.
Functional annotation of retained SNPs revealed that the majority were situated in intergenic regions (51.63%) or intronic regions (37.30%). Exonic SNPs, which accounted for 0.76% of the total SNPs (Figure 1A), included 102,215 missense SNPs, 145,226 synonymous SNPs, 1213 stop-gained SNPs, 278 stop-lost SNPs, 253 start-lost SNPs, 131 start/stop-retained SNPs, and 22 initiator-codon SNPs (Additional file 3). For the InDels, functional annotation demonstrated that the majority were also located in intergenic regions (50.31%) or intronic regions (38.60%). Exonic InDels accounted for 0.14% of the total InDels (Figure 1B), comprising 935 frameshift-insertion InDels, 1460 frameshift-deletion InDels, 590 inframe-insertion InDels, 807 inframe-deletion InDels, 40 stop-gained InDels, 34 stop-lost InDels, 23 start-lost InDels, 11 start/stop-retained InDels, 9 gene-fusion InDels, and 1 exon-loss InDel (Additional file 4).Figure 1Functional classification of the detected SNPs and InDels in genomes. A Frequency of different functional types of the total SNPs. B Frequency of different functional types of the total InDels.
Differential variants between carriers and noncarriers
To elucidate the genetic underpinnings of susceptibility to the FMDV carrier state, we focused on variants showing marked differences in allele frequency between carriers and noncarriers. These variants are hypothesized to play a critical role in the pathogenesis of persistent FMDV infection. Considering the potential influence of population structure on the analysis results, we performed principal component analysis (PCA) and kinship analysis. The results revealed no significant population stratification between carriers and noncarriers (Additional files 5A, 5B, 6). Subsequently, we performed a case–control (noncarrier–carrier) GWAS using the R package rMVP with GLM, MLM, and FarmCPU models simultaneously. GLM model identified six autosomal SNPs supposed to be significantly associated with the FMDV carrier state, with five SNPs in the intergenic regions and one in the intron (Additional files 7A, 7B, 8). Unexpectedly, only one locus (NC_037337.1:32701) was observed in carriers, which was more prevalent in noncarriers. Notably, no variants were found to be significantly associated with the carrier status in MLM and FarmCPU models. This outcome is highly probable owing to the inadequate sample size.
To address this, we applied Fisher’s exact test to identify variants exhibiting statistically significant differences in allele frequency between the two groups. We identified 532 exonic SNPs showing extremely significant differences in allele frequency between carriers and noncarriers (*p-*value < 0.01). These SNPs were located within 286 genes, including 523 missense SNPs in 278 genes, 4 start-lost SNPs in 4 genes, 4 stop-gained SNPs in 3 genes, and 1 stop-lost SNP in 1 gene (Additional file 9). Notably, the majority of these differential SNPs were located predominantly on chromosomes 8, 16, and 22 (Figure 2).Figure 2Distribution of the differential SNPs (p-value < 0.01) between carrier and noncarrier groups identified by Fisher’s exact test. The x-axis represents the chromosome positions and the y-axis indicates the chromosome number.
Concurrently, we identified 22 extrenely differential exonic InDels (p-value < 0.01) between carriers and noncarriers that covered 21 genes, including 7 inframe-deletion InDels within 7 genes, 5 i-frame-insertion InDels within 5 genes, 4 frameshift-deletion InDels within 4 genes, 4 frameshift-insertion InDels within 4 genes, 1 stop-gained InDel within 1 gene, and 1 stop-lost InDel within 1 gene (Additional file 10). These differential InDels were distributed mainly on chromosomes 2, 8, 10, 11, 16, 18, 22, 25, 27, and 29 (Figure 3A). Among them, the largest one was a 15-bp insertion, while the majority ranged from 1 to 3 bp (Figure 3B).Figure 3Distribution of differential InDels (p-value < 0.01) between carrier and noncarrier groups identified by Fisher’s exact test. A The number of differential InDels across chromosomes. B Distribution of differential InDels length (bp).
Variants with highly allelic frequency differentiation between carriers and noncarriers
We then used the fixation index (FST) method to quantify each missense variant across autosomal exonic regions and splicing donor sites to identify variants with extreme allele frequency differentiation between carrier and noncarrier groups. On the basis of the top 1% of the FST values, we identified 1063 SNPs covering 660 genes (FST ≥ 0.242161, Figure 4A; Additional file 11) and 15 InDels covering 14 genes (FST ≥ 0.257986, Figure 4B; Additional file 12). These variants represent the most significant differences in allele frequency between carrier and noncarrier groups.Figure 4Genome-wide scans for signatures of selection between carrier and noncarrier groups. A Manhattan plot for the FST values (y-axis) of selected autosomal SNPs across all autosomes (x-axis). B Manhattan plot for the FST values (y-axis) of selected autosomal InDels across all autosomes (x-axis). The dashed lines represent thresholds for the top 1% FST values.
Candidate variants associated with the carrier state
To confirm genetic variations significantly associated with persistent FMDV infection, we intersected genomic regions meeting two stringent criteria: (1) p-values < 0.01 based on Fisher’s exact test, and (2) FST values within the top 1% of the distribution. In total, we identified 384 overlapping SNPs covering 224 genes and 7 overlapping InDels covering 7 genes (Figure 5). Among these overlapping regions, we further identified 49 carrier-specific variants (Additional file 13). These variants encompassed 40 genes, including 6 variants covering 2 interferon-stimulated GTPase genes (1 variant within LOC101903126, 5 variants within LOC783920), 5 variants covering 3 GTPase IMAP family genes (2 variants within GIMAP8 and LOC768255, respectively, and 1 variant within LOC512867), 2 variants covering 2 genes related to antigen processing and presentation (CTSL, BLA-DQB), 2 variants covering 2 genes related to natural killer (NK) cell activity (NKTR, KIR2DS1), 4 variants covering 4 olfactory receptor family genes (LOC783328, LOC104968693, LOC788663, OR2W1), 2 variants covering 2 genes related to chromatin structure and histone methylation (RBBP5, HIST1H2AC_1), 2 variants covering 2 transcription factor genes (ZBTB47, ZNF205), 12 variants covering 12 genes related to cell proliferation, differentiation and cytoskeleton remodeling (CYLC2, IRX3, DCT, LOC100299102, TRANK1, SPTA1, EFCAB13, MEGF9, EGFL7, DCDC2C, LOC529969, MOS), and 14 variants covering 11 genes related to diverse signal transduction (1 variant within IL5RA, LOC112443721, LOC788915, ADAT1, NUBPL, BPIFA4, LOC101906978, and SNTG2 repectively; 2 variants within* LOC522174*, LOC534913, and FRRS1L repectively).Figure 5Identification of the overlapping variants. Venn diagram showing the overlapping region counts of SNPs (left) and InDels (right) obtained based on Fisher’s exact test and FST methods.
Cattle–yak haplotype assemblies mapping
To assess potential artefacts arising from hybrid recombination, the clean reads were mapped to cattle–yak (Bos taurus × Bos grunniens) haplotype 1 and haplotype 2 assemblies after quality filtering. The average genome coverage was greater than 97% (Additional file 14). PCA analysis revealed distinct mutation patterns separating FMDV carriers from noncarriers as well as animal individuals (Additional file 15). We then mapped the identified candidate variants based on the cattle (Bos taurus) reference genome (ARS-USD1.2) to two cattle–yak (Bos taurus × Bos grunniens) haplotype genomes. Among 49 variants identified on the basis of the ARS-USD1.2 genome, 24 variants were consistently present across both cattle–yak haplotype genomes, including 3 variants within 3 olfactory receptor family genes (LOC783328, LOC104968693, LOC788663), 1 variant within 1 gene involved in antigen processing and presentation (CTSL), 2 variants covering 2 genes related to NK cell activity (NKTR, KIR2DS1), 1 variant within 1 gene involved in chromatin structure and histone methylation (HIST1H2AC_1), 9 variants covering 9 genes related to cell proliferation, differentiation and cytoskeleton remodeling (CYLC2, IRX3, DCT, TRANK1, SPTA1, MEGF9, DCDC2C, LOC529969, EFCAB13), and 8 variants covering 8 genes related to signal transduction (IL5RA, ADAT1, LOC101906978, LOC534915, BPIFA4, FRRS1L, NUBPL, SNTG2) (Additional file 16). These results suggest that the prevalence of these 24 variants is independent of hybrid recombination and instead associated solely with the carrier status.
Subsequently, we determined 31 additional differential carrier-specific variants restricted to cattle–yak haplotype genomes (Additional file 17). Notably, 5 additional variants have been identified within 4 olfactory receptor genes (2 variants within LOC618052; 1 variant within LOC618140, LOC100297284,and LOC784614 respectively), suggesting that olfactory receptor genes may play a significant role in FMDV colonization in the nasopharynx. The other variants included 4 variants within 4 genes related to regulation of gene transcription and translation (ZNF74, ZNF114, ZNF213, TCOF1), 2 variants within 2 genes related to energy metabolism (MDH1B, LOC519145), 5 variants within 5 genes related to cell development and morphological maintenance (CEP120, DCDC2C, IRX6, NEK5, LAMB3), 7 variants within 7 genes related to stress resistance (SEC31A, PAQR4, LOC100139663, LOC404051, ENDOV, GOLGA5, DNAJC4), 4 variants within 4 genes related to regulation of immune cell function (LOC508459, RGMB, INPP5D, SIGLEC11), and 4 variants within 4 genes related to diverse transduction (LOC101906978, TRIM15, CACNA2D3, LOC517901). Intriguingly, no single variant was present in all carrier individuals, with multiple distinct mutations coexisting within each individual. This observation suggests that the establishment of persistent FMDV infection is governed by the concerted action of multiple genes rather than being driven by a single genetic factor.
Discussion
In this study, we conducted whole-genome resequencing on carrier and noncarrier individuals to identify genetic factors associated with the carrier status. Based on the comparative analyses using the cattle (Bos taurus) reference genome (ARS-USD1.2) and cattle–yak (Bos taurus × Bos grunniens) haplotype genomes, we found that significantly differential carrier-specific variants were located in genes encoding olfactory receptors (LOC783328, LOC104968693, LOC788663, LOC618140*, LOC100297284, LOC618052, *LOC784614), as well as genes involved in regulating cell development and morphological maintenance (CYLC2, IRX3, DCT, TRANK1, SPTA1, MEGF9, DCDC2C, LOC529969, EFCAB13, CEP120, IRX6, NEK5, LAMB3), transcription and translation regulation (HIST1H2AC_1, ZNF74, ZNF114, ZNF213, TCOF1), signal transduction (IL5RA, LOC101906978, LOC534915, BPIFA4, FRRS1L, SNTG2, TRIM15, CACNA2D3, LOC517901), energy metabolism (MDH1B, LOC519145), stress resistance (ADAT1, NUBPL, SEC31A, PAQR4, LOC100139663, LOC404051, ENDOV, GOLGA5, DNAJC4), and immune cell function (LOC508459, RGMB, INPP5D, SIGLEC11, CTSL, NKTR, KIR2DS1). These genetic variants potentially determine the innate immune regulatory capacity and the magnitude of adaptive immune activation, thereby conferring distinct antiviral capacities.
Olfactory receptors (ORs) belong to the class A G protein-coupled receptors and constitute the most important chemosensory receptor family responsible for the sense of smell in the nasal olfactory epithelium. The bovine nasopharyngeal mucosal epithelium serves as a critical viral sanctuary for both primary and persistent FMDV infection [7], indicating a potential link between ORs and FMDV infection. Nasal ORs, which are expressed in olfactory sensory neurons, are critical for olfactory cognition and associated neuronal plasticity. The olfactory route has been shown to facilitate the entry of neurotropic viruses, such as poliovirus, herpesviruses, Japanese encephalitis virus, and influenza A virus [52], into the central nervous system. In cattle, FMDV is mainly transmitted through the respiratory tract. Consequently, mutations in OR genes may facilitate viral colonization of the nasopharynx and contribute to viral persistence in carriers.
Transcriptional regulation of genes governs diverse antiviral responses. HIST1H2AC_1 encodes replication-dependent histones, which are highly expressed just before the S-phase and are then repressed after DNA replication [53]. Mutations in HIST1H2AC_1 have contributed to carcinogenesis [54]. ZNF74, ZNF114, and ZNF21 belong to an evolutionarily conserved but poorly explored Krueppel C2H2-type ZNF family, and their transcriptional repression by their KRAB domain is critical for early embryonic development [55]. ZNF74 is a developmentally expressed and commonly deleted gene in DiGeorge syndrome characterized by thymic hypoplasia/aplasia and hypoparathyroidism [56]. ZNF114 interacts with TRIM28 to control self-renewal of human pluripotent stem cells through epigenetic repression of prodifferentiation genes [57]. ZNF213 has been demonstrated to negatively control breast cancer progression [58]. In addition, signal transduction is also crucial for regulating gene expression. Our comprehensive analysis revealed 9 genes (IL5RA, LOC101906978, LOC534915, BPIFA4, FRRS1L, SNTG2, TRIM15, CACNA2D3, LOC517901) that are functionally associated with these processes. For example, CACNA2D3 encodes a subunit of voltage‐dependent calcium channels and is crucial in calcium signaling, impacting immune cell functions and inflammatory responses. CACNA2D3 variants have been shown to correlate with asthma and atopic dermatitis multimorbidity in children [59]. TRIM15 is a TNF-α-induced late response gene and inhibits the TNF-α-induced NF-κB pathway, thereby acting as a feedback modulator to keep the proinflammatory NF-κB pathway under control [60]. Knockdown of TRIM15 limited virus/RIG-I ligand-induced interferon production and enhanced vesicular stomatitis virus replication [61]. Consequently, genetic variations in these genes may contribute to individual differences in animal development and defense responses to viral infection, influencing the outcomes of viral infections.
FMDV is a prototypical cytolytic virus. Upon infection, FMDV rapidly hijacks the host biosynthetic machinery to facilitate its replication and induces host cell lysis to enable subsequent rounds of infection. Studies have demonstrated that FMDV infection triggers robust cellular stress responses, including cytoskeletal reorganization [62], cell cycle arrest [63], metabolic disturbance [64], endoplasmic reticulum stress [65, 66], autophagy [66, 67], and apoptosis [68]. These processes contribute to the suppression of viral spread and the activation of antiviral immune defenses aimed at viral clearance. Our study identified a catalog of variants in the carrier individuals, which is located in genes involved in regulating cell development and morphological maintenance (CYLC2, IRX3, DCT, TRANK1, SPTA1, MEGF9, DCDC2C, LOC529969, EFCAB13, CEP120, IRX6, NEK5, LAMB3), energy metabolism (MDH1B, LOC519145), and stress resistance (ADAT1, NUBPL, SEC31A, PAQR4, LOC100139663, LOC404051, ENDOV, GOLGA5, DNAJC4). While these genes have not been documented in FMDV-related research, accumulating evidence indicates that their proper function plays a pivotal role in controlling pathogen infections. For instances, variants in DCDC2C were associated with susceptibility to salmonella in pig [69]; SPTA1 mutations were significantly associated with hepatitis D viral infection [70]. NEK5 interacts with mitochondrial proteins and interferes negatively in mitochondrial mediated cell death [71]; in Chlamydia trachomatis infection, GOLGA5 plays a critical role in maintaining the structural integrity of the Golgi apparatus [72]. Therefore, genetic mutations in key components of these biological processes potentially compromise host's intrinsic defense mechanisms, leading to FMDV persistence.
In general, innate immunity primarily functions by secreting cytokines to recruit and activate immune cells, and adaptive immunity is responsible for the ultimate clearance of viral infections. An optimal immune response is characterized by efficient antigen presentation as well as strong and well-balanced humoral and cellular immunity. Here, we identified seven genetic variants in genes involved in regulating antigen presentation (CTSL), NK (NKTR, KIR2DS1) and T (LOC508459, RGMB, INPP5D, SIGLEC11) cell function. Cathepsin L (CTSL) is involved in MHC II-dependent immune responses by cleaving the chaperone molecule invariant chain, which supports MHC II assembly in the endoplasmic reticulum into intermediate products [73]. Accordingly, functional mutations in CSTL affect FMDV antigen presentation and immune cell activation. NK cells play critical roles in innate antiviral immunity by recognizing and lysing virus-infected cells. A lack of or deficiency in NK cell function has been demonstrated to lead to increased susceptibility to viral infection [74]. NKTR and KIR2DS1 are important receptors that regulate NK cell cytotoxicity. Genetic variants in KIR2DS1 are associated with increased susceptibility to HCV infection in a high-risk Chinese population [75]. In contrast to the suppressed NK activity observed in FMDV-infected swine, NK cells play a role in host immune responses against FMDV in cattle [76]. Moreover, studies have shown that the effector functions of INPP5D-deficient NK and T cells are compromised in vivo [77]. Collectively, the variations in genes involved in regulating immune cell function potentially impair antiviral cellular immune activation, thereby facilitating viral persistence.
While our screened genes did not overlap with those previously reported in transcriptomic datasets from tissues of FMDV persistently infected individuals [35, 36, 78], they are all involved in comparable biological processes that are intimately linked to the host's antiviral defenses and self-protection mechanisms. Mutations in these genes may impair the host's antiviral response and self-repair capacity, thereby facilitating long-term viral persistence. Another possible explanation is that genes showing marked changes in transcriptomic levels are generally downstream effector genes, whereas most of the variants we identified are located in key genes that regulate these downstream effectors.
In conclusion, our study comprehensively evaluated genetic variants associated with the carrier status through WGS analyses. These findings provide novel insights into the genetic architecture underlying persistence and offer avenues for improved prevention and control strategies. Although we investigated susceptibility to persistent FMDV infection using high-throughput WGS, we were aware of the limitations of this study, such as the small sample size and the absence of interspecies and intersex comparisons. Constraints of laboratory capacity and eradication policies make it difficult to obtain large numbers of samples from animals with established persistent infections.
Supplementary Information
Additional file 1. **Basic information of enrolled subjects.**Additional file 2. **Summary of whole genome sequences mapped to the cattle reference genome.**Additional file 3. **Distribution of SNPs identified in cattle individuals within various genomic regions based on the cattle reference genome.**Additional file 4. Distribution of Indels identified in cattle individuals within various genomic regions based on the cattle reference genome.Additional file 5. Population genetic structure. A Principal component analysisof the distribution of all genotyped SNPs from all resequencing samples. B Kinship of all resequencing samples. CR, carriers; NCR, noncarriers.Additional file 6. Kinship analysis of all subjected animals based on the cattle reference genome.Additional file 7. Genome-wide association study. A Quantile–quantile plot of GWAS using GLM, MLM, and FarmCPU tests. B Manhattan plots showing SNPs significantly associated with the carrier state. The X-axis represents the chromosome, and the Y-axis indicates −log_10. **Dots over the dotted line indicate significantly associated SNPs.Additional file 8. Variants significantly associated with the FMDV carrier state identified by a GLM model based on the cattle reference genome.Additional file 9. Differential missense SNPs with p-value < 0.01 of Fisher’s exact test between carriers and noncarriers based on the cattle reference genome.**Additional file 10. **Differential missense Indels with p-value < 0.01 of Fisher’s exact test between carriers and noncarriers based on the cattle reference genome.**Additional file 11. **SNPs with the top 1% **FST_ **values represent extreme allele frequency differences between carriers and noncarriers based on the cattle reference genome.**Additional file 12. Indels with the top 1% F_ST_ **values represent extreme allele frequency differences between carriers and noncarriers based on the cattle reference genome.**Additional file 13. **Genetic variations exclusively identified in carrier individuals based on the cattle reference genome.**Additional file 14. **Summary of whole genome sequences mapped to cattle–yak haplotype genomes.**Additional file 15. **Principal component analysis of the SNP distribution from all resequencing samples.**A The PCA representation of haplotype genomes 1 and 2 separately. B **The overlay representation of PCA based on haplotype genomes 1 and 2.**Additional file 16. Genetic variations extensively identified in carrier individuals based on the cattle reference genome and cattle–yak haplotype genomes.Additional file 17. Genetic variations exclusively identified in carrier individuals based on cattle–yak haplotype genomes.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1The genome sequence archive in the National Genomics Data Center, China National Center for Bioinformation. https://ngdc.cncb.ac.cn/gsa