Sex-specific gonadal transcriptome during early development of Siberian sturgeon
André Lasalle Gerla, Germán Benech-Correa, Christophe Klopp, Denise Vizziano-Cantonnet

TL;DR
This study identifies sex-specific genes in early gonad development of Siberian sturgeon, revealing new candidates for male and female differentiation.
Contribution
The first transcriptomic analysis of undifferentiated gonads in genetically sexed Siberian sturgeon reveals both established and newly identified genes activated near the onset of sex differentiation.
Findings
Female-biased genes include estrogen-related genes and novel candidates not previously linked to estradiol.
Male differentiation involves the known gene tbx1 and new candidates like plin1 and nrxn3.
No sex-biased genes related to androgen production were identified in males.
Abstract
Sex determination and differentiation are complex processes shaped by a wide variety of molecular factors. In contrast to teleost species, many aspects of these processes remain poorly understood in basal non-teleost fishes such as the Siberian sturgeon (Acipenser baerii). Genetic sexing of this important aquaculture species now enables studies of undifferentiated males and females to identify factors involved in early sexual differentiation. Twelve undifferentiated Siberian sturgeon (six males, six females) were genetically sexed at 2.5 months of age. High-quality RNA was extracted from gonad samples, and transcriptomes were assembled using a reference dataset. Bioinformatic analyses were performed to identify sex-biased genes through differential expression analysis, Gene Ontology (GO) enriched terms, and classification of coding and non-coding sequences. Genes potentially…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
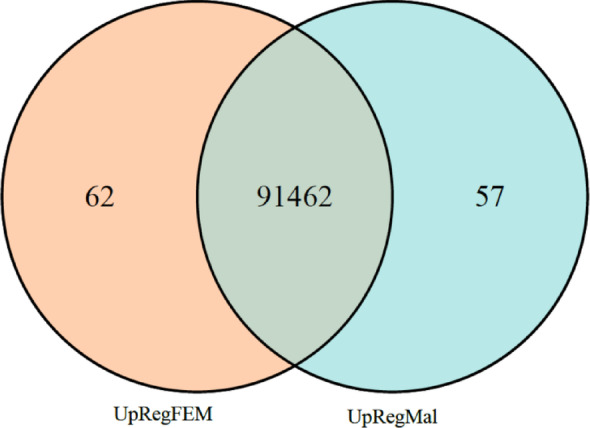 Figure 1
Figure 1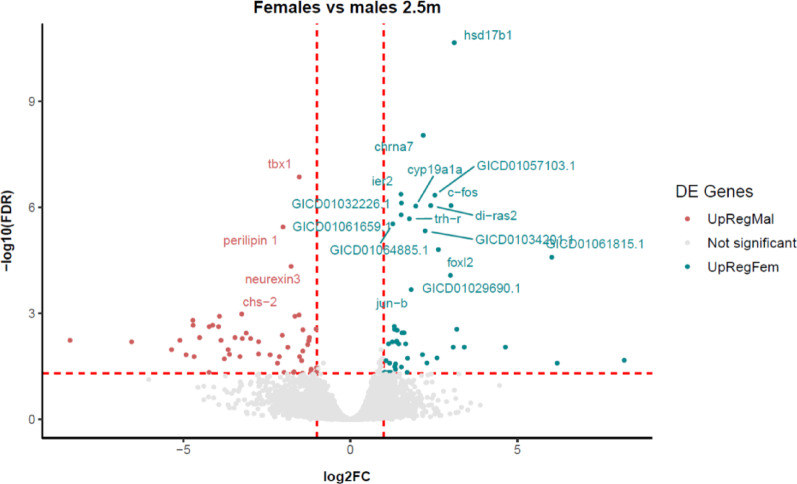 Figure 2
Figure 2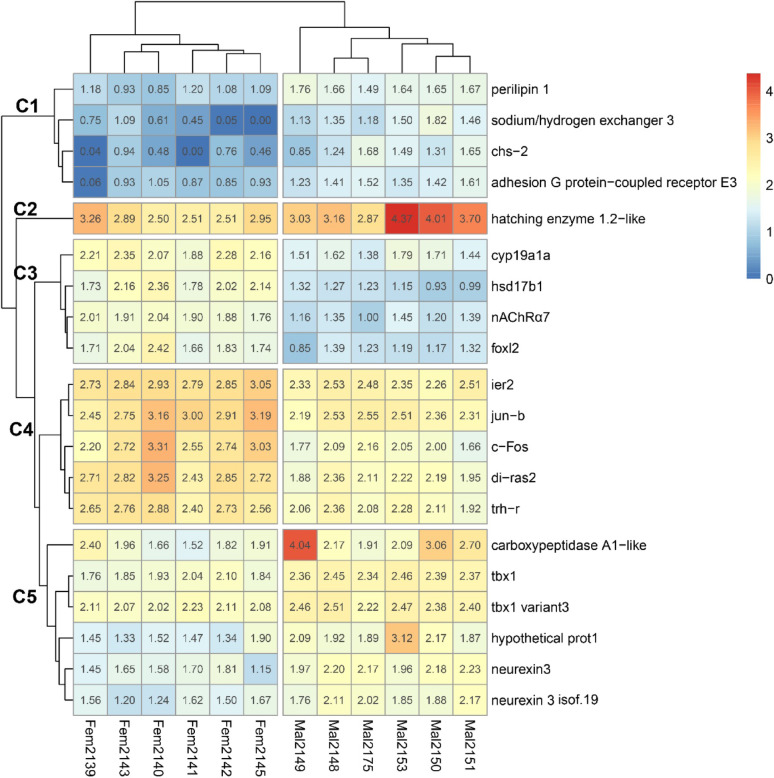 Figure 3
Figure 3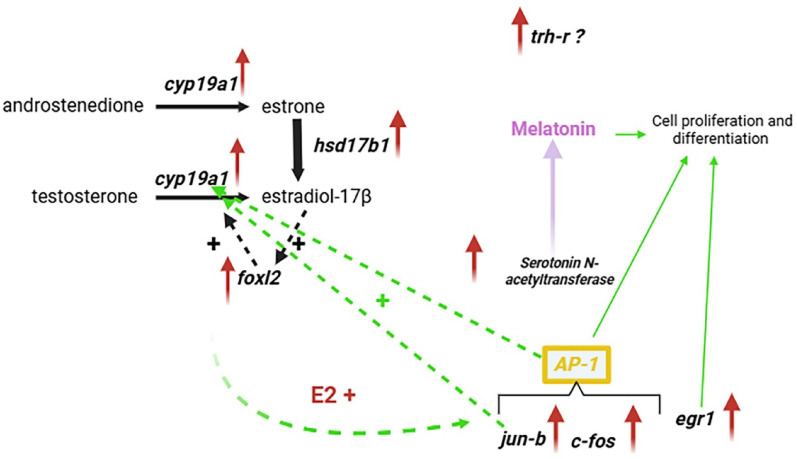 Figure 4
Figure 4- —https://doi.org/10.13039/501100006049Comisión Sectorial de Investigación Científica
- —https://doi.org/10.13039/100008725Agencia Nacional de Investigación e Innovación
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenetic and Clinical Aspects of Sex Determination and Chromosomal Abnormalities · Reproductive biology and impacts on aquatic species · Developmental Biology and Gene Regulation
Background
Sex determination and differentiation are highly complex biological processes that have been extensively studied in teleost species, but their mechanisms remain poorly understood in basal fishes such as the Siberian sturgeon (Acipenser baerii). Studying these processes in slow-developing fish like sturgeons offers a valuable opportunity to investigate conserved and divergent mechanisms of sex regulation in vertebrates.
In teleosts, sex determination can involve genetic, environmental, or mixed mechanisms. Several master sex-determining genes have been described [1, 2], but most are not conserved across species. In contrast, key genes involved in estrogens synthesis (cyp19a1, foxl2, hsd17b1) are broadly conserved among teleosts [3–7], supporting the view that estrogens are major inducers of sex differentiation in fish [8]. In the non-teleost Siberian sturgeon, these three genes are also activated very early in female gonads [9], months before the first signs of sexual differentiation appear [10].
Other genes such as Wnt family member 4 (Wnt4), Beta catenin (Ctnnb1), and R-spondin 1 (Rspo1), crucial in the female pathway in mammals [11], are also involved in sex differentiation in some fish [7]. However, these genes do not show sex-dimorphic expression in Siberian sturgeon during early molecular differentiation [9]. Similarly, in the male pathway, genes such as anti-Müllerian hormone (amh), doublesex and mab-3 related transcription factor 1 (dmrt1), SRY-box transcription factor 9 (sox9), and desert hedgehog (dhh) are well known to promote testis differentiation in many teleosts [6, 12] do not show sex-specific expression at this stage in Siberian sturgeon [9]. In contrast, gonadal soma-derived factor (gsdf), a gene expressed in teleost gonads [13–16] and involved in male differentiation [17–19], has been detected in Siberian sturgeon databases [20], and one copy is activated in the male program [21].
Like many other sturgeons, Siberian sturgeon exhibit relatively slow gonadal development and very late puberty [22]. Sexual differentiation occurs in the juvenile stage, at around 5–6 months of age [10, 23], several months after gonad formation at ~ 1 month. This extended window provides an opportunity to study molecular differentiation prior to morphological changes.
Because sexual differentiation involves multiple cell types and extensive cell–cell communication, we hypothesized that additional, as yet undescribed, genes contribute to early differentiation in sturgeon. To test this hypothesis, we performed transcriptomic analyses of undifferentiated gonads from 12 genetically sexed individuals (six males, six females) at 2.5 months of age, that is, 1.5 months after gonad formation and coinciding with the onset of molecular differentiation [9]. This approach allowed us to identify sex-biased expression patterns and candidate genes and molecular pathways that could contribute to the control of early sexual differentiation.
Methods
Fish maintenance and sampling
All experiments and animal maintenance were carried out in accordance with the Comisión Honoraria de Experimentación Animal (CHEA) protocol N° 1516 (exp. 240012-000023-22). Siberian sturgeon was provided by the Estuario del Plata sturgeon farm (Tacuarembó, Uruguay) and transported to the Facultad de Ciencias (Universidad de la República Oriental del Uruguay, Montevideo) aquaculture facilities at two months of age. Fish were maintained under natural photoperiod conditions, in a 500 L tank with 250 L of daily water renewal, at natural spring temperatures (17–21 °C). Fish were fed daily at 2% of body weight. Fin clips were collected at two months of age and preserved in 95% ethanol (PPA) until DNA extraction for sexing by PCR. Fish were then acclimated for 15 days before gonad sampling, performed on 12 genetically sexed males and females. Length (± 1 mm) and weight (± 0.01 g) were recorded (Additional File 1).
Genomic DNA and RNA extraction
Genomic DNA extraction was performed following the laboratory protocol. Each Siberian sturgeon caudal fin sample (2 mm) was placed in a sterile 1.5 mL microcentrifuge tube. Twenty-five microliters of tissue digestion buffer (25 mM NaOH and 0.2 mM EDTA) were added to each sample and vortexed briefly. Samples were then heated for 15 min at 95 °C in a BIOER LifeECO thermocycler, vortexed again, and centrifuged. Following digestion, 25 µL of neutralization buffer (40 mM Tris, pH 8) was added. DNA samples were stored at − 20 °C until use. Total RNA extraction was performed immediately after gonad sampling using the Monarch RNA extraction kit (New England Biolabs), following the manufacturer’s instructions.
Sexing and RNA sample selection
Fish were sexed by PCR with gDNA using the genetic sex marker published by Kuhl et al. [24]. Each reaction contained 0.625 µL of 10 µM forward and reverse primers, 20 ng of gDNA, 0.125 µL of NZYTech polymerase (Thermo Fisher Scientific, Waltham, MA, USA), 2.5 µL of 10× NZY Taq buffer, and 3.75 µL of 2 mM dNTP mix (Thermo Fisher Scientific, Waltham, MA, USA), with nuclease-free water added to a final volume of 25 µL. The thermal cycler program was: 95 °C for 3 min; 35 cycles of 95 °C for 30 s, 56 °C for 30 s, and 72 °C for 45 s; followed by a final extension at 72 °C for 8 min. Sex was assessed by analyzing the PCR product on a 2% agarose gel as described by Kuhl et al. [24].
Female and male gonadal RNA samples from genetically sexed fish were identified, and 6 samples of each sex were selected for RNA-Seq analysis. RNA quality was assessed using an Agilent Bioanalyzer 2100. RNA integrity numbers (RIN) between 8 and 10 indicated high-quality RNA in the selected samples (Additional File 2).
RNA-Seq
All RNA sequencing was performed by Macrogen (Korea). RNA integrity was confirmed using the TapeStation RNA ScreenTape system prior to library construction. Libraries were prepared using the Illumina TruSeq Stranded Total RNA kit with Ribo-Zero Gold. Sequencing was performed on a NovaSeq 6000 platform, generating 100 bp paired-end reads, with approximately 40 million reads per sample, corresponding to roughly 4 Gb of raw data (in FASTQ format).
Gonad transcriptome assembly and annotation
The transcriptome was assembled using the Siberian sturgeon gonadal transcriptome [20] as a reference. Raw read files were processed with fastp v0.23.4 [25] using default parameters to remove adapters and low-quality bases. Cleaned reads were then aligned with BWA-MEM v0.7.17 [26] against the GICD01.1 Siberian sturgeon reference transcriptome available in the Transcriptome Shotgun Assembly (TSA) database at NCBI. Alignments were sorted, compressed, and indexed with SAMtools v1.x [27] using the sort, view, and index functions with default parameters. Expression levels were quantified with SAMtools idxstats, and expression tables were generated using standard Unix cut and paste commands.
Differential gene expression analysis
Female and male samples were compared. Each gene was quantified as fragments per kilobase of transcript per million mapped reads (FPKM). Differential expression analysis between the two sexes was performed in R v4.3.1 using the Bioconductor package edgeR. A false discovery rate (FDR) < 0.05 was used as the threshold for defining differentially expressed genes (DEGs). Genes meeting this criterion were considered differentially expressed. The functions calcNormFactors, estimateCommonDisp, estimateTagwiseDisp, and exactTest were applied with default parameters, as described in the edgeR user guide. DEGs overexpressed in females and males were analyzed separately for Gene Ontology (GO) enrichment.
Identification of protein-coding and non-coding RNA
Female- and male-biased DEG nucleotide sequences were translated using the ExPASy Translate tool (https://web.expasy.org/translate/) to determine which of them encoded proteins. The amino acid sequences of predicted coding ORFs (open reading frames) were compared against public databases using BLASTp (NCBI) to assign gene identities. If no protein-coding ORF was detected, the contig was classified as “non-coding RNA” [28]. Non-coding RNAs were not further analyzed due to the lack of a published Siberian sturgeon genome.
GO enrichment analysis
(GO) enrichment was performed using topGO [29] (http://bioconductor.org/) with the classic algorithm and Fisher’s exact test. For each list of differentially expressed genes (DEGs), enrichment was assessed for Biological Process (BP), Molecular Function (MF), and Cellular Component (CC) categories.
All DEGs were mapped to GO terms in the Gene Ontology database, and statistical enrichment analysis was performed. GO terms with an adjusted p < 0.05 were considered significantly enriched (Additional Files 3 and 4).
Statistical analysis
The assumptions of normality and homogeneity of variance were evaluated for length and weight data and for expression-level comparisons between clusters. When these assumptions were met, Student’s t-test was applied; if the assumptions were not met, the Mann–Whitney test was used [30]. Statistical significance was set at p < 0.05.
Results
Differential expression between female and male gonads
No differences in length or weight were detected between males and females used for transcriptomic analysis (t-test, p > 0.05).
The assembled gonadal transcriptome of female and male Siberian sturgeon 2.5 months of age produced 91,581 contigs. Of these, 119 were differentially expressed between sexes (FDR < 0.05): 0.07% (n = 62) were upregulated in females and 0.06% (n = 57) in males (Fig. 1, Additional Files 5 and 6). The remaining 91,462 contigs showed no significant differences. Only 32 female-biased and 29 male-biased contigs encoded proteins (Additional Files 7 and 8). The remaining contigs corresponded to overexpressed non-coding RNA (Additional Files 5 and 6).
Fig. 1. Venn diagram representing the 62 upregulated contigs in females (FDR < 0.05) (UpRegFEM); 57 upregulated contigs in males (FDR < 0.05) (UpRegMal); and 91,462 non-significant contigs (FDR > 0.05)
Volcano plot analysis of upregulated sequences
Figure 2 shows that hsd17b1 and tbx1 were the most significantly upregulated genes in females and males, respectively. The results were markedly more significant for hsd17b1 than for other female genes (FDR = 2.18E^− 11^), such as cyp19a1 (FDR = 9.17E^− 07^) or foxl2 (FDR = 1.57E^− 05^). In males, tbx1 (FDR = 1.38E^− 07^) was most significantly upregulated, followed by perilipin 1 (plin1, 3.59E^− 06^) and neurexin 3 (nrxn3, 4.69E^− 05^).
Fig. 2. Volcano plot representation of the common contigs (grey) and contigs upregulated in females (turquoise) and males (red). Dotted lines indicate arbitrary thresholds for reference
Heatmap representation of differentially expressed genes
The top 20 coding contigs were separated into five clusters according to expression pattern similarity (Fig. 3). Cluster 1 (C1) included plin1, slc9a3 (sodium/hydrogen exchanger 3), chs2-like (chitin synthase 2-like), and adgre3 (adhesion G protein-coupled receptor E3), all of which were male-biased. Cluster 2 (C2) consisted of a single male-biased gene encoding the hatching enzyme 1.2-like protein.
Clusters 3 (C3) and 4 (C4) contained female-biased genes. C3 grouped together hsd17b1, cyp19a1, foxl2, and chrna7 (alpha-7 nicotinic cholinergic receptor subunit). C4 included the immediate early response gene ier2, transcription factors such as jun-b and c-fos, signaling factors such as di-ras2 (Ras-related protein), and hormone-related genes such as trh-r (thyrotropin-releasing hormone receptor). Between the female-biased clusters, genes in C4 showed significantly higher expression than those in C3 (Shapiro–Wilk p = 0.09; Levene p = 0.02; Mann–Whitney p = 0.005).
C5 contained the male-biased genes tbx1 and nrxn3, along with one isoform (neurexin 3 isoform 19). It also included carboxypeptidase A1-like and a hypothetical protein-coding gene with an open reading frame (ORF) that has not yet been characterized in available databases.
In addition to the genes represented in the heatmap, other genes were found to be overexpressed in female gonads. Among them, serotonin N-acetyltransferase (aanat), a key enzyme in melatonin biosynthesis, and the one coding for the transcription factor Egr1 (early growth response 1) stand out as particularly.
Fig. 3. Heatmap of the top 20 codifying contigs. Each line corresponds to one contig and each column to one sample from male (Mal) or female (Fem) groups. The scale from 0 to 4 corresponds to level of contig expression (Z-score). C1 to C5 are the five different clusters
Gene ontology (GO) enrichment
GO analysis of female-upregulated contigs (n = 62) revealed 186 Biological Processes (BP), three Cellular Components (CC), and six Molecular Function (MF) GO terms that were significantly enriched (adjusted p < 0.05) (Additional File 3). In males, the Gene Ontology analysis of upregulated contigs (n = 57) identified 256 enriched GO terms in the BP category, 7 in CC, and 23 in MF (p adjusted p < 0.05) (Additional File 4).
From the list of enriched GO terms, we selected those associated with gonadal processes related to sex differentiation and maturation in females and males (Additional Files 9 and 10). Notable GO terms identified in females included “steroid hormone biosynthesis,” “gonadotropin response” (FSH, LH), “granulosa cell differentiation,” “melatonin metabolism” and “transcription factors such as AP-1” (Additional File 3 and 9). In males, “hormone metabolic process” and “retinoic acid signaling” were among the most relevant differentially enriched GO terms (Additional File 10).
Discussion
This study presents the first transcriptomic analysis of undifferentiated gonads from genetically sexed Siberian sturgeon during early molecular sex differentiation, at 2.5 months of age [9]. This approach provides a snapshot of this complex process during a critical developmental period, pointing to gene and pathway networks that may drive gonadal differentiation [9, 31]. Gene expression profiles were largely conserved between the sexes, with 99.87% of the studied contigs showing no expression differences between males and females. Early female and male sexual differentiation, therefore, is likely guided by the 0.13% of the total genes expressed in gonads showing sex-biased activation (0.07% in future ovaries, 0.06% in future testis). Most of the genes are likely crucial for the survival and proliferation of gonadal cells, as observed in mammals [11].
Validation of the gonadal transcriptome
The transcriptome was validated by the identification of known female-specific genes (e.g., cyp19a1, foxl2, hsd17b1) among the top differentially expressed contigs [9, 31]. These results are consistent with our previous qPCR studies with and without sex markers [9, 31–33]. Moreover, the crucial role of estradiol-17β in Siberian sturgeon ovarian differentiation has been reported during phenotypic male-to-female sex reversal treatments with exogenous E2 [10]. These validations based on previous work [9, 10] strengthens these new results and supports the involvement of the newly identified overexpressed genes in female and male sexual differentiation pathways, including their involvement from early stages in this complex process.
Novel candidates in sex differentiation
New gene candidates for sex differentiation emerged from the heatmap clustering analysis. Estrogens-related genes (hsd17b1, cyp19a1, foxl2) were grouped in Cluster 3, while Cluster 4 included other genes overexpressed in female gonads such as jun-b, c-fos, ier2, di-ras2, and trh-r. The uniform expression levels within each cluster suggest the presence of synexpression groups of genes that could be co-expressed and potentially co-regulated, indicating that these genes may function together within common regulatory pathways to reach a common goal, in this case to drive sex-specific development.
Many of the aforementioned genes may be estrogens-responsive or act downstream of estrogen signaling. Immediate early genes such as jun-b and c-fos are well known to be rapidly stimulated by estrogens [34, 35]. Although not included in the heatmap, egr1 was significantly upregulated in females in our differential expression analysis. egr1 is expressed in somatic cells of the ovary and testis and can be stimulated by 17β-estradiol [36–38]. This gene functions as a transcription factor regulating cellular growth and differentiation in response to mitogenic and stress signals [39–41]. In Acanthopagrus latus, a hermaphroditic fish, egr1 is overexpressed in the female pituitary during the transition from an ovotestis to a definitive ovary, consistent with a role in female differentiation [42]. In mammals, Egr1 expression varies with the estrous cycle, and its overexpression influences brain plasticity and female-specific behaviors [43], further supporting its importance in female-related pathways.
Although little is known about their regulation during ovarian differentiation, the expression of ier2 has been shown to be stimulated by E2 in the rat uterus [44], and the presence of di-ras2 in Cluster 4 suggests that this gene could also be modulated by estrogens in the gonadal differentiation context.
Finally, trh-r also appears to be estrogen-sensitive, as estrogens upregulate TRH receptor expression in mammotropic cells [45–47]. Thus, we suggest that genes in Cluster 4 are part of an estrogens-driven synexpression group, in which functionally related genes are coordinately expressed in response to estrogens produced by genes in Cluster 3 (hsd17b1, cyp19a1, foxl2).
Functional overview of the genes in cluster 4
jun-b and c-fos emerge as particularly relevant due to their established roles as immediate early response genes [48, 49]. These genes encode transcription factors that dimerize to form the AP-1 complex, which can regulate downstream targets including cyp19a1 [50, 51], thereby indirectly influencing estrogens production. The AP-1 complex, as well as jun and fos individually, are broadly involved in cell proliferation and differentiation [49, 52–54].
Evidence from other species supports their potential role in ovarian development: c-fos has been implicated in ovary formation in Drosophila [55], while in chickens, silencing of jun causes phenotypic masculinization by repressing the classical female genes foxl2,* cyp19a1*, and esr1 and upregulating male pathway genes such as dmrt1 and sox9 [56]. To date, neither jun-b nor c-fos have been reported in the context of gonadal differentiation in fish; nevertheless, our findings suggest that they may play key regulatory roles in early ovarian differentiation in Siberian sturgeon.
ier2 encodes a transcription factor that belongs to the immediate early gene group. This gene is a downstream target of fibroblast growth factor (FGF) signaling that plays a role in the establishment of left-right asymmetry and convergent extension movements in zebrafish [57] and Xenopus [58]. It is known that FGF signaling acts on multiple types of somatic cells within the developing gonad in the larval and pupal stages of Drosophila and is necessary for maintaining fertility [59]. In mammals Ier2 expression has been detected in the ovary [60] and, more specifically, in the rat uterus, Arao et al. [44] demonstrated that the gene is stimulated by 17β-estradiol (E2).
di-ras2 acts as a tumor suppressor by inhibiting the Wnt/β-catenin pathway [61, 62], which regulates expression of genes involved in cell proliferation, differentiation, and maintenance across species and plays a well-established role in gonadal development in mammals [11]. Although di-ras2 has not been directly associated with gonadal differentiation, we hypothesize that it may influence the proliferation of specific cell types stimulated by estrogens. This effect could be mediated through pathways regulated by immediate early genes such as jun-b and c-fos, which can act individually or as AP-1 dimers.
Activation of trh-r in female gonads contrasts with the typical association of the thyroid axis with male differentiation [63]. In Xenopus laevis larvae, inhibition of TSH leads to feminization [64, 65], while in zebrafish, administration of thyroid hormones promotes testis development by upregulating male-associated and downregulating female-associated genes [66]. In medaka (Oryzias latipes) and rice field eel (Monopterus albus), thyroid hormone treatment likewise activates the male differentiation pathway and stimulates the production of 11-oxygenated androgens [67, 68].
These findings suggest that the thyroid axis may function differently in Siberian sturgeon, possibly in a species-specific manner. One possibility is that TRH is produced by the ovary itself and acts in an autocrine or paracrine fashion to regulate female gonadal development. However, our previous tissue and gonadal transcriptome analyses [20, 69] showed that trh is highly expressed in the brain but not in the gonads of either sex at any developmental stage. This raises the question of whether hypotalamic-produced TRH circulates to act on gonadal trh-r at 2.5 months of age, or whether TRH receptors instead interact with another locally produced factor that activates the female pathway.
Interestingly, the female-biased genes in Cluster 4, which have received less attention in the literature in the context of sex differentiation, showed significantly higher expression than those in Cluster 3, which contains the classic estrogens-production genes (hsd17b1,* cyp19a1*, and foxl2). This finding highlights their potential importance in the early development and regulation of female gonads and suggests that these two gene clusters may interact during the initial stages of gonadal differentiation.
Novel hormonal players in ovarian differentiation
The discovery of serotonin N-acetyltransferase overexpression and enrichment of the GO term “melatonin metabolism” is particularly novel in the context of ovarian sex differentiation. Melatonin has traditionally been linked to circadian rhythms and reproductive processes, and it is produced in both male and female gonadal tissues [70, 71]. In Cyprinus carpio, treatment with aromatase inhibitors during early sex differentiation to induce female-to-male transdifferentiation is accompanied by reduced estradiol and melatonin levels, suggesting a cooperative role of these hormones in ovarian differentiation [72]. These findings raise important questions about whether melatonin acts synergistically with estradiol in promoting female development, or whether it can influence ovarian differentiation independently. Further hormone administration studies will be required to clarify this point.
Ovarian regulatory model near the onset of sex differentiation
As we previously demonstrated [9, 10], future ovaries are capable of producing estrogens by 2.5 months of age. This capacity is supported by activation of the enzymes hsd17b1 and cyp19a1 (aromatase), which are essential for estrogens biosynthesis, and of the transcription factor foxl2, which stimulates aromatase expression.
The present study confirms the early estrogen production pathway and identifies new estrogens targets, such as immediate early genes jun-b, c-fos, and egr-1 (Fig. 4). c-fos, jun-b, and egr-1 are activated downstream, stimulated by estrogen signaling. jun-b can stimulate aromatase either independently or by forming a dimer with c-fos, which acts as a transcription factor called AP1. We propose a new positive feedback loop for aromatase stimulation through jun-b and AP1, independent of foxl2 (Fig. 4, dashed green arrows).
Fig. 4. Proposed regulatory model for early ovarian sex differentiation in Siberian sturgeon. Red arrows indicate upregulated genes. Thick solid black arrows represent well-characterized regulatory pathways. Dashed black arrows indicate known positive regulatory interactions. Thin solid green arrows denote established functions involving factors activated in this study. Dashed green arrows represent potential positive regulatory interactions among components. Purple highlights the proposed production of melatonin. A question mark (?) indicates the uncertain role of trh-r
In turn, AP-1, formed by the products of jun-b and c-fos, together with egr1, may regulate the cell proliferation and differentiation processes required for development of the female gonad (Fig. 4, solid green arrows). This proposed network consolidates the estrogenic identity of the ovary near the onset of molecular sexual differentiation, with a gene node downstream of E2 capable of two major functions: maintaining ovarian estrogens levels through stimulation of aromatase, and regulating cell proliferation and differentiation.
Moreover, the potential involvement of hormones such as melatonin and TRH, which may act synergistically with estrogens (Fig. 4), adds further complexity to early ovarian differentiation in fish and other vertebrates. This underscores the importance of newly identified sex-differentiation-related genes in the earliest stages of ovarian development.
Potential role of non-coding RNAs in gonadal differentiation
The overexpression of non-coding transcripts in both male and female gonads suggests the involvement of additional regulatory pathways of gene transcription. In fish, long non-coding RNAs can directly interact with chromatin and regulate transcription through epigenetic modifications [73]. Epigenetic regulation of sex-differentiation genes has been reported in Cynoglossus semilaevis and linked to sexual differentiation [74, 75]. It is therefore possible that some of the non-coding transcripts identified here act as epigenetic factors influencing gene transcription during early sex differentiation in Acipenser baerii. However, without a reference genome, we cannot definitively classify these contigs as long non-coding RNAs, and such analyses will require new genome assemblies. Nevertheless, this represents a promising direction for future genomic and epigenetic research in sturgeons.
Perspectives on testicular differentiation: early candidates in a delayed process
The early male differentiation pathway remains poorly understood. Here, 29 contigs with coding ORFs were overexpressed in males. Unlike the ovarian molecular signature, male gonads at this early timepoint lacked the classical genes typically involved in testicular differentiation, consistent with observations at more advanced stages in previous studies [9, 31]. Notably, this is the first report of tbx1, a gene previously linked to testicular differentiation in trout, being overexpressed at this early stage of development [76, 77]. Moreover, tbx1 was the most strongly expressed of all male-biased genes, representing a promising early marker of testicular differentiation.
tbx1 also has well-established roles in male gonadal fate across vertebrates. Functional evidence from amphibians and reptiles [78–80] shows that tbx1 represses the classical ovarian pathway (e.g., cyp19a1,* foxl2*) while stimulating testicular development (e.g., amh,* dmrt1*,* wt1*,* cyp26a*). In Andrias davidianus, feminizing estradiol treatments repress tbx1 expression [78]. Interestingly, at this early stage of development, no genes related to androgen production were detected in future testes, suggesting that these factors are not directly involved in early male gonadal differentiation, an idea previously proposed by Lasalle et al. [9]. Taken together, these findings indicate that in an estrogen-free microenvironment that creates the conditions for male differentiation, tbx1 may act as an early upstream regulator of testicular development in A. baerii.
mmp9, another important male upregulated gene, encodes a metalloproteinase involved in extracellular matrix remodeling, a process critical for testis formation [81] and germ cell migration [82–84]. In sturgeon, metalloproteinases have previously been linked to gonadal development [31]. A similar role has been described in the protogynous species Synbranchus marmoratus, where metalloproteinases facilitate ovarian regression and testicular development [85]. Together, the available evidence suggests that mmp9 may play a structural and organizational role in testicular morphogenesis in sturgeon, enabling germ cell migration during the early stages of testicular differentiation.
Other significantly male-skewed genes remain functionally uncharacterized, possibly reflecting unknown sex-specific functions before or during testicular differentiation. Studying these factors will be essential for completing our understanding of the male differentiation network.
Sex-specific GO term enrichment
The functional pathways derived from the GO analysis reflect the progression of sex differentiation in males and females. The two sexes showed distinct patterns of enriched terms. The differentially activated GO terms in females were closely associated with sex differentiation, determination, and hormone production (i.e. estrogens), along with metabolic processes in the gonads. In contrast, fewer of the differentially expressed GO terms in males appeared to be linked to sex-specific processes, and instead were mainly associated with retinoic acid and general hormone metabolism. None of the male-biased GO terms were related to androgen production or receptivity [86, 87]. In females, the abundance of GO terms directly associated with gonadal processes indicates a more advanced gonadal development at this stage compared to the male process. The pattern of male and female GO terms suggests that sex differentiation progresses more slowly in males than females, consistent with results in Siberian sturgeon in later stages of gonad development [31].
Conclusion
Our results support the idea that molecular sexual differentiation begins by 2.5 months of age and is more advanced in females than males at this stage.
This first transcriptomic analysis of undifferentiated gonads reveals the activation of both classic estrogens-production genes and novel female-biased candidates, including immediate early response genes as well as genes not previously linked to estrogen signaling. The established male factor tbx1 was overexpressed in males, along with previously unrecognized candidates. Furthermore, as in prior studies, no sex-biased genes related to androgen production were detected, suggesting that early male gonadal development may proceed independently of steroid production.
Overall, this exploratory study highlights the complexity of pathways involved in gonadal differentiation and proposes new hypotheses regarding the mechanisms and factors that may operate during the early stages of gonadal development in basal fish.
Supplementary Information
Supplementary Material 1
Supplementary Material 2
Supplementary Material 3
Supplementary Material 4
Supplementary Material 5
Supplementary Material 6
Supplementary Material 7
Supplementary Material 8
Supplementary Material 9
Supplementary Material 10
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Klopp C, Lasalle A, Di Landro S, Vizziano-Cantonnet D. RNA-Seq transcriptome data of undifferentiated and differentiated gonads of Siberian sturgeon. Data Brief. 2020;31:105741. 10.1016/j.dib.2020.105741 PMC 728075032529009 · doi ↗ · pubmed ↗
- 2Benech-Correa G, Lasalle Gerla AE, Vizziano Cantonnet D. gsdf is the only major pro-male gene activated during the molecular sex differentiation period of Siberian sturgeon. 30th CECE & 9th ISFE Joint Conference of the European Society for Comparative Endocrinology and of the International Society for Fish Endocrinology; Faro, Portugal. 2022.
- 3Fujimoto J, Hori M, Ichigo S, Morishita S, Tamaya T. Estrogen induces expression of c-fos and c-jun via activation of protein kinase C in an endometrial cancer cell line and fibroblasts derived from human uterine endometrium. Gynecol Endocrinol. 1996;10:109 – 18.10.3109/095135996090979008701784 · doi ↗ · pubmed ↗
- 4Webb DK, Moulton BC, Khan SA. Estrogen induces expression of c-jun and jun-b protooncogenes in specific rat uterine cells. Endocrinology. 1993;133:20 – 8.10.1210/endo.133.1.83195688319568 · doi ↗ · pubmed ↗
- 5Punetha M, Kumar S, Paul A, Jose B, Bharati J, Sonwane A, et al. Deciphering the functional role of EGR 1 in prostaglandin F 2 alpha induced luteal regression applying CRISPR in corpus luteum of Buffalo. Biol Res. 2021;54:9. 10.1186/s 40659-021-00333-7PMC 795360933712084 · doi ↗ · pubmed ↗
- 6Feng K, Su J, Wu Z, Su S, Yao W. Molecular cloning and expression analysis of Thyrotropin-Releasing Hormone, and its possible role in gonadal differentiation in rice field eel Monopterus albus. Animals. 2022;12:1691. 10.3390/ani 12131691 PMC 926498435804589 · doi ↗ · pubmed ↗
- 7Klopp C, Cabau C, Greif G, Lasalle A, Di Landro S, Vizziano-Cantonnet DJD. Siberian sturgeon multi-tissue reference transcriptome database. Database. 2020;2020.10.1093/database/baaa 082PMC 768768033238003 · doi ↗ · pubmed ↗
- 8Zhang Y, Xu F, Li H, Chen S. Differential methylation reveals pathways associated with sex differentiation in Chinese tongue sole (Cynoglossus semilaevis). Aquaculture and Fisheries; 2024.