Heterocyclizations at the Isocyanide Carbon: Mechanistic Insights into the 2‑Isocyanoaniline to Benzimidazole Conversion
Mateo Alajarin, Marta Marin-Luna

TL;DR
This paper explores how a specific chemical conversion from isocyanoaniline to benzimidazole occurs, revealing a possible new reaction pathway.
Contribution
The study provides a computational analysis of a previously unclear reaction mechanism involving isocyanoaniline cyclization.
Findings
A transient 1,4-diazabutatriene intermediate is proposed to lower the reaction barrier in the cyclization.
The reaction pathway involving the intermediate is more plausible than a unimolecular concerted process.
Abstract
Benzo-fused heteroazacycles can form through spontaneous cyclizations of phenylisocyanides bearing some electron donor substituents at the ortho position. Although such reactions have been known since the 1970s, a clear mechanistic understanding of these transformations remains absent. Here, we disclose a detailed computational analysis aimed at elucidating the more plausible mechanism for one of these cyclizations, the o-isocyanoaniline to benzimidazole conversion. Our results show that a transient 1,4-diazabutatriene could play a key role in this cyclization by enabling a reaction pathway of lower barrier than the unimolecular concerted process.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
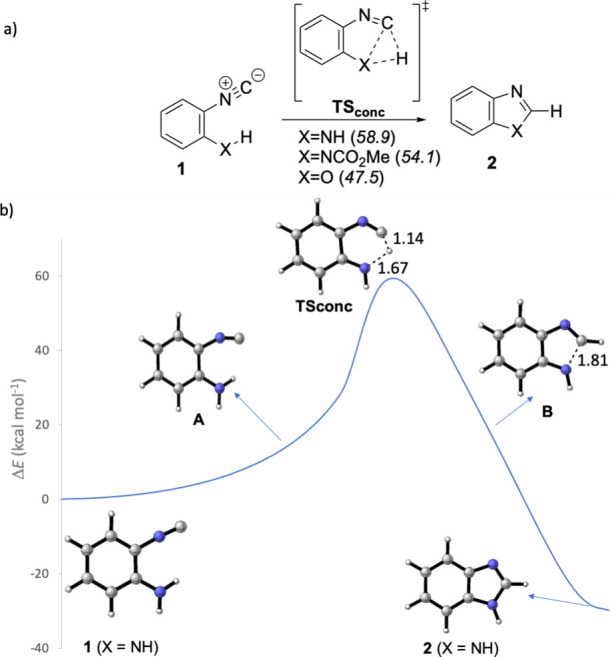 Figure 1
Figure 1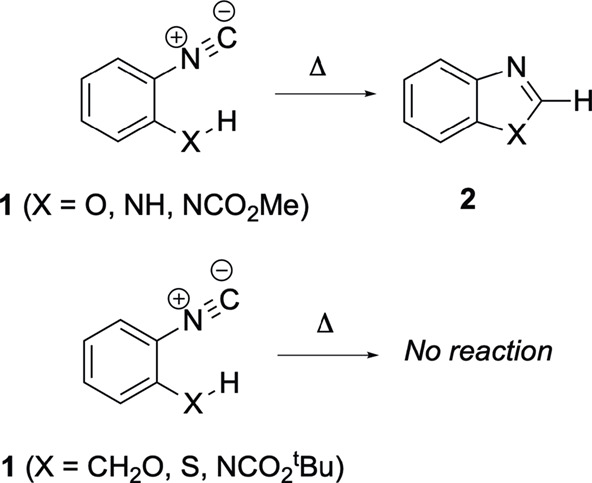 Figure 2
Figure 2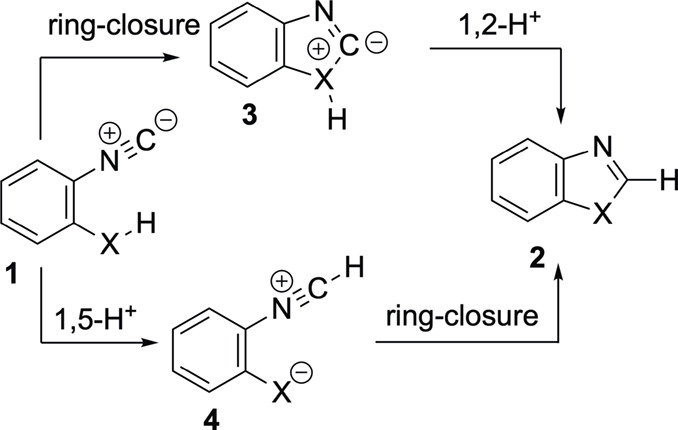 Figure 3
Figure 3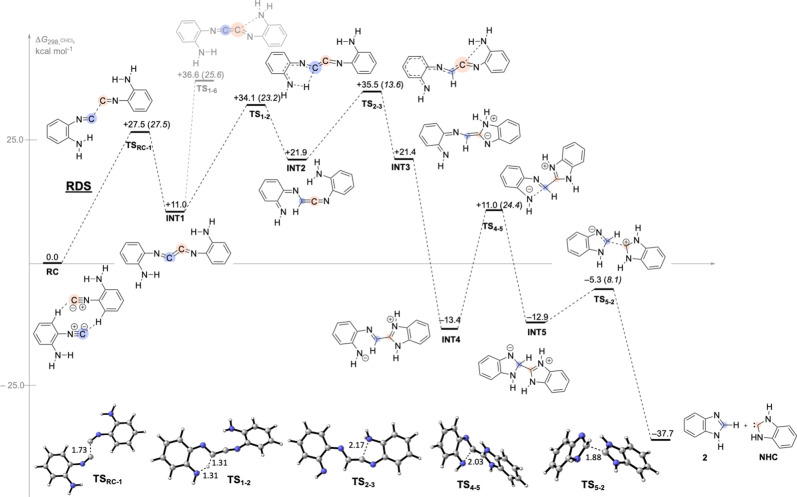 Figure 4
Figure 4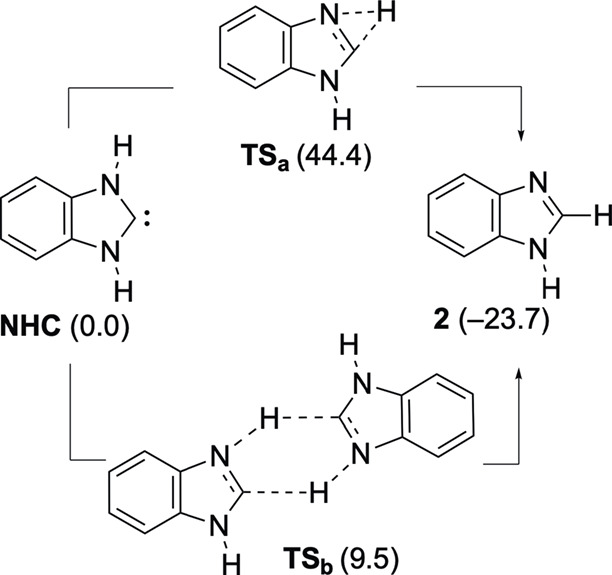 Figure 5
Figure 5 Figure 6
Figure 6 Figure 7
Figure 7- —Fundaci?n S?neca10.13039/100007801
- —Ministerio de Ciencia, Innovaci?n y Universidades10.13039/100014440
- —European Regional Development Fund10.13039/501100008530
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCatalytic C–H Functionalization Methods · Synthesis and Reactivity of Heterocycles · Synthesis of heterocyclic compounds
Isocyanides (isonitriles) are versatile functional groups that take part in multiple organic transformations either as nucleophiles or electrophiles.? Due to such remarkable dual reactivity, isocyanides have come to be known as chameleonic compounds. ?−? ? ? Among other things, isocyanides are capable of forming complex azacyclic compounds via multicomponent reactions.? In this sense, the formation of benzo-fused heterocycles 2, starting from ortho-substituted phenylisocyanide fragments 1 (X = O, NH), is an interesting reported protocol (Scheme). o-Isocyanophenol evolves to benzoxazole upon thermal treatment, ?−? ? ? ? and its amino partner has been described as an unstable species due to its rapid evolution to benzimidazole.? It is noteworthy that the success of these transformations is contingent upon the direct bonding of the heteroatom to the aromatic ring. In contrast, o-(hydroxymethyl)phenyl isocyanide exhibits stability under thermal conditions.? Reverse reactions, from benzo-fused azacycles toward o-substituted phenylisocyanides, have also been reported, proceeding under photochemical activation. ?,? Curiously, while the conversion of benzothiazole into o-isocyanobenzenethiol is a known reaction, there is no experimental evidence substantiating the inverse transformation, from isocyano-thiol to benzothiazole.?
Other types of phenylisocyanides ortho-decorated with N-acyl functions seem to exhibit analogous transformations. While tert-butyl (2-isocyanophenyl)carbamate has been reported to be stable under thermal reaction conditions,? other 2-isocyanophenyl carbamates undergo, when warmed up, an intramolecular cyclization to give the corresponding 1-acylbenzimidazoles (Scheme). ?−? ?
While these are recognized transformations, to our knowledge, their respective mechanisms have not been clarified. Our recent research in the chemistry of azido-substituted 2-(heteroaryl)phenyl isocyanides ?,? has led us to focus our attention on the cyclizations summarized in Scheme and their intriguing mechanisms. Herein, we disclose the results of our computational scrutiny of such mechanisms at the PCM(CHCl_3_)/M06-2x/aug-CC-pvTZ//M06-2x/aug-CC-pvDZ theoretical level, concluding with the probable participation of 1,4-diazabutatrienic dimers of the starting ortho-substituted phenylisocyanides.
We first proposed the two-step process starting with the formation of an X–C bond in 1, leading to the heterocyclic ylide 3, followed by a 1,2-H shift, where H migrates as a proton, from the XH fragment to the carbanionic C atom, finally giving rise to the benzoheterocycle 2. However, all attempts to locate either the transition structures corresponding to the cyclization first step or the cyclized ylide 3 failed, including those incorporating explicit solvent molecules in the calculations (Scheme). On the other hand, Reva et al. proposed that the transformation of 2-isocyanophenol 1 (X = O) into benzoxazole occurs by an alternative two-step mechanism, involving the initial 1,5-H shift from the OH group toward the C atom of the isocyanide at 1, leading to the conjugated nitrile ylide 4 (X = O) that further collapses into the respective 2 (Scheme).? Curiously, these authors apparently neither computed the first step, the 1,5-H shift, nor showed the transition structure of the second step, the respective final cyclization of 4 to 2. They present only a relaxed PES-scan of this latter step as the only support of their mechanistic proposal. Probably, they could not locate the two transition structures of this two-step process. Indeed, this was also our case after many unsuccessful attempts.
Notwithstanding, our calculations show that the three scrutinized 1 to 2 cyclizations (X = O, NH, NCO_2_Me) could occur in a concerted polar manner (Figurea). Our DFT calculations predict that the amino-isocyanide must pay an energy penalty of 58.9 kcal mol^–1^ to evolve toward benzimidazole 2. In the case of carbamate, the computed free energy barrier is 54.1 kcal mol^–1^. The lowest, although still high, computed free energy barrier is associated with the formation of benzoxazole (47.5 kcal mol^–1^). These concerted processes were ascertained by the intrinsic reaction coordinate analyses performed on the located transition structures TS _ conc _ (Figureb for model X = NH). The respective single steps start by the rehybridization of the sp-N atom of the isocyanide moiety to sp^2^, thus placing the C atom well oriented to abstract the H atom of the XH group (A in Figureb) while moving toward the transition structure TS _ conc _. In the second part of the reaction coordinate, the residual NH group is then capable of linking to the C atom of the isocyanide, thus forming the final five-membered ring (B in Figureb). With these results in hand, we do not consider these concerted mechanisms as plausible because the computed high activation energies seem not compatible with the reported smooth reaction conditions (room temperature).
After discarding the three unimolecular mechanisms above, we next tested some bimolecular scenarios, as a transformation mediated by a second isocyanide molecule acting as a kind of catalyst in a total concerted mechanism (see the Supporting Information). Unfortunately, we cannot validate that proposal since the key transition structure was not located.
The experimental evidence that bulky (2-isocyanophenyl) carbamates are stable under thermal conditions led us to consider if the 1 → 2 conversion is due to the delocalization of the X atom lone pair on the aromatic system, rather than to the acidity of the XH proton or the nucleophilic character of X.
For this reason, and following a logical approach, we next gave a chance to see a stepwise version of the bimolecular mechanistic proposal commented above. We conceived such process as initiated by the formation of a dimer of the ortho-HX-phenyl isocyanide molecules. In 1965, Ugi et al. already proposed that phenylisocyanide is in equilibrium with the dimer formed by linking the isocyano carbon atoms of two molecules by means of a double bond.? In fact, such dimers, the elusive 1,4-diazabutatrienes, have been envisaged as intervening in multiple transformations of isocyanides although they have been never isolated.?
Taking as computational model 1 (X = NH) and admitting its initial dimerization to 1,4-diazabutatriene INT1 (Scheme), two routes emerge henceforth depending on how the carbon atoms of the NCCN fragment act either as nucleophilic, by the transfer of one proton of the amino group to the C atom, or as electrophilic, by the attack of the amino group on the C atom. Afterward, each route could branch off into secondary pathways toward benzimidazole 2. All of these routes have been computed (see the Supporting Information), and we will show here only our results on the most favorable pathways in energy terms (Scheme).
Two molecules of o-aminophenyl isocyanide interact with each other to form a reactant complex RC,? whose structural organization provides an optimal scenario to a successful head-to-head dimerization process, forming the 1,4-diazabutatriene intermediate INT1 (ΔG ^‡^ TS _ RC‑1 _ = 27.5 kcal mol^–1^). This dimerization step is computed as the rate-determining step (RDS) of the entire process. This step was also explored on 2-isocyanophenol 1 (X = O) and on the carbamate derivative 1 (X = NCO_2_Me). Pleasantly, our calculations predict that the activation energies of both processes are lower than those of the previously computed concerted mechanisms: 26.3 and 27.5 kcal mol^–1^, respectively.
Next, INT1 could experiment the nucleophilic attack of the amino N atom to the nearest cumulated carbon by surpassing an energy barrier of 25.6 kcal mol^–1^ via TS _ 1–6 _. However, we found a lower energetic pathway with a barrier of 23.2 kcal mol^–1^, converting INT1 into the ketenimine intermediate INT2, via TS _ 1–2 _, by a 1,5-H transfer from the amino group to the nearest cumulated C atom. The difference between the activation barriers of the latter two processes could be reasonably interpreted. In the case of the nucleophilic attack of the amino N atom, the structural distortion needed to reach the cumulated carbon atom is notable, whereas in the 1,5-H step, the migrating H atom is already in the vicinity of the recipient C atom. After that, the ketenimine fragment of INT2 is prone to experiment the classical nucleophilic attack of the pendant amino group to its central carbon atom (ΔG ^‡^ TS _ 2–3 _ = 13.6 kcal mol^–1^), yielding INT3, in a proton-transfer equilibrium with INT4 (−13.4 kcal mol^–1^). Note here the low barrier of the nucleophilic addition leading to INT3 compared to that surpassing TS _ 1–6 _. This fact might be a consequence of the unequivocal electrophilic character of the central C atom of the ketenimine function, similar to that of INT2. A subsequent ring closure at INT4 via TS _ 4–5 _ (ΔG ^‡^ TS _ 4–5 _ = 24.4 kcal mol^–1^) affords the zwitterionic structure INT5. The low-energy rupture of the exocyclic C–C bond results in the formation of benzimidazole 2 and the carbene NHC.?
The last step of our mechanistic proposal consists of the transformation of NHC into its more stable isomer benzimidazole 2 (Scheme). Although the much higher stability of 2 in comparison with NHC has been disclosed in advance by Alkorta and Elguero,? to our knowledge, the precise mechanism of the conversion of NHC into 2 has not been computed until now. We have found that the intramolecular 1,2-H transfer via TS _ a _ is nearly 35 kcal mol^–1^ more energetically costly than the bimolecular version through the 6-center transition structure TS _ b _ (Scheme). These findings agree with those described for other N-heterocyclic carbenes.?
In summary, herein, we disclose a detailed computational study on the transformation of o-isocyanoaniline into benzimidazole, a transformation briefly described in the bibliography but not mechanistically scrutinized up to now. Multiple reaction pathways have been computed, and the results show that the most favorable one involves the dimerization of the reactant to form a 1,4-diazabutatriene intermediate. This step continues with a proton transfer from the amino group to one of the cumulated carbon atoms of that intermediate, followed by two cyclizations and the dissociation into two molecules of benzimidazole. This mechanism has been computed to be more favorable, in energy terms, than the unimolecular one previously proposed, which we computed to occur by a concerted mechanism with a high energy barrier.
A relevant conclusion of this work is that mechanistic pathways involving the transient formation of dimeric 1,4-diazabutatrienes should be taken into consideration in the study of the mechanism of other reported cyclizations involving the carbon atom of isocyanide functions. This could be the case for the Van Leusen reaction and related 5-endo-dig cyclizations. Computational insights on this theme are currently underway in our research group.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Moderhack D.N-Isocyanides-Synthesis and Reactions Tetrahedron 201268305949596710.1016/j.tet.2012.04.099 · doi ↗
- 2Gomes G. D. P.Loginova Y.Vatsadze S. Z.Alabugin I. V.Isonitriles as Stereoelectronic Chameleons: The Donor–Acceptor Dichotomy in Radical Additions J. Am. Chem. Soc.201814043142721428810.1021/jacs.8b 0851330270623 · doi ↗ · pubmed ↗
- 3Vatsadze S. Z.Loginova Y. D.dos Passos Gomes G.Alabugin I. V.Stereoelectronic Chameleons: The Donor–Acceptor Dichotomy of Functional Groups Chem. – Eur. J.201723143225324510.1002/chem.20160349127862399 · doi ↗ · pubmed ↗
- 4Marin-Luna M.Alajarin M.In Search of 1,4-Diazabutatrienes, the Elusive Isocyanide Homodimers: The Superchameleonic F–NCJ. Org. Chem.20208518119751197910.1021/acs.joc.0c 0168532840374 · doi ↗ · pubmed ↗
- 5Marin-Luna M.Alajarin M.Unraveling the Computed Non-Least Motion Pathway for the Homodimerization of Superchameleonic Isocyanides: The Peculiar Nonsymmetrical (F–NC) 2 Reactant Complex Phys. Chem. Chem. Phys.20212331169731698010.1039/D 1CP 02674 G 34338701 · doi ↗ · pubmed ↗
- 6Dömling A.Ugi I.Multicomponent Reactions with Isocyanides Angew. Chem., Int. Ed.200039183168321010.1002/1521-3773(20000915)39:18<3168::AID-ANIE 3168>3.0.CO;2-U 11028061 · doi ↗ · pubmed ↗
- 7Ferris J. P.Antonucci F. R.Trimmer R. W.Mechanism of the Photoisomerization of Isoxazoles and 2-Cyanophenol to Oxazoles J. Am. Chem. Soc.197395391992010.1021/ja 00784 a 048 · doi ↗
- 8Reva I.Jesus A. J. L.Nunes C. M.Roque J. P. L.Fausto R.UV-Induced Photochemistry of 1,3-Benzoxazole, 2-Isocyanophenol, and 2-Cyanophenol Isolated in Low-Temperature Ar Matrixes J. Org. Chem.20218696126613710.1021/acs.joc.0c 0297033872502 · doi ↗ · pubmed ↗