Chemical Synthesis of Pseudomonas aeruginosa, Staphylococcus aureus, and Acinetobacter baumannii Capsular Polysaccharide Fragments as Leads for Cross‐Protection
Amar Kumar Mishra, Emelie E. Reuber, Diksha Rai, Leif E. Sander, Julia Duerr, Simon Y. Graeber, Marcus A. Mall, Bettina C. Fries, Peter H. Seeberger, Suvarn S. Kulkarni

TL;DR
This paper describes the chemical synthesis of bacterial sugar fragments that could lead to vaccines offering protection against multiple drug-resistant pathogens.
Contribution
The study introduces a novel chemical synthesis approach to create cross-protective epitopes from three MDR pathogens.
Findings
Three conserved epitopes with strong cross-reactive immunogenicity against P. aeruginosa, S. aureus, and A. baumannii were identified.
A trisaccharide was found to be the minimal epitope required for cross-protective immune responses.
Efficient synthesis strategies were developed for key glycan structures relevant to vaccine development.
Abstract
Pseudomonas aeruginosa and Staphylococcus aureus are listed by the World Health Organization as high‐priority multidrug‐resistant (MDR) pathogens, whereas Acinetobacter baumannii is classified as the critical‐priority group. These bacteria cause life‐threatening infections such as severe bloodstream, nosocomial, urinary tract, and soft‐tissue infections. Their cell surfaces display complex and structurally distinct glycans absent in host cells, making them targets for glycoconjugate vaccine and diagnostic research. In this study, we report the chemical synthesis of mono‐ and oligosaccharide fragments derived from three ESKAPE pathogens, P. aeruginosa O11, S. aureus (CP5, CP8, and strain M), and A. baumannii (S34 and O5), as well as Plesiomonas shigelloides O1. Glycan microarray screening revealed three epitopes exhibiting strong cross‐reactive immunogenicity against P. aeruginosa, S.…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
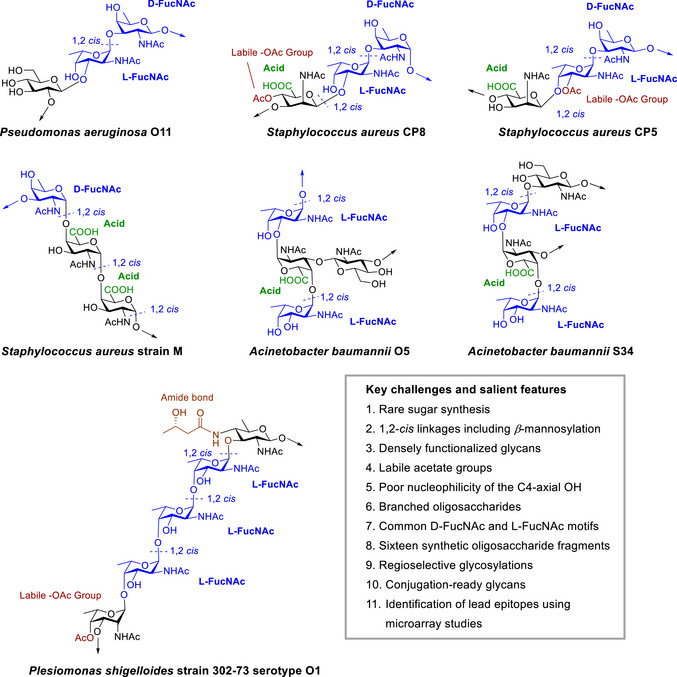 Figure 1
Figure 1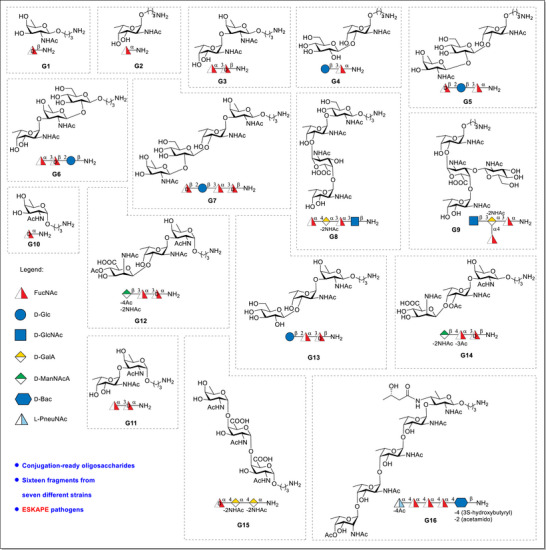 Figure 2
Figure 2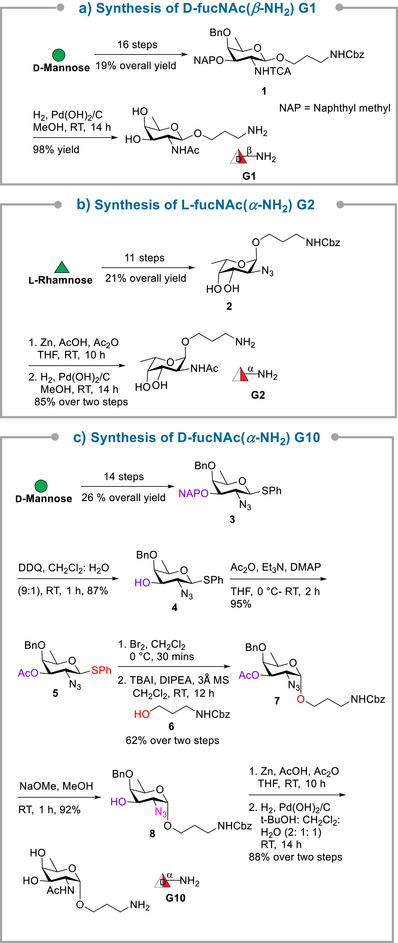 Scheme 1
Scheme 1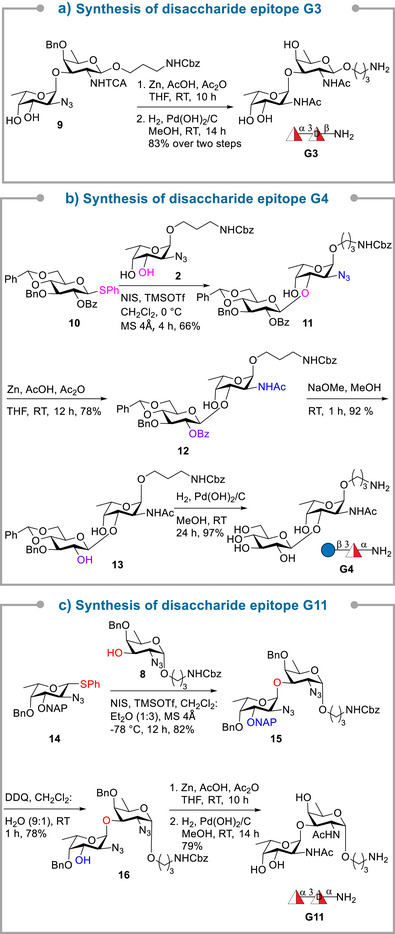 Scheme 2
Scheme 2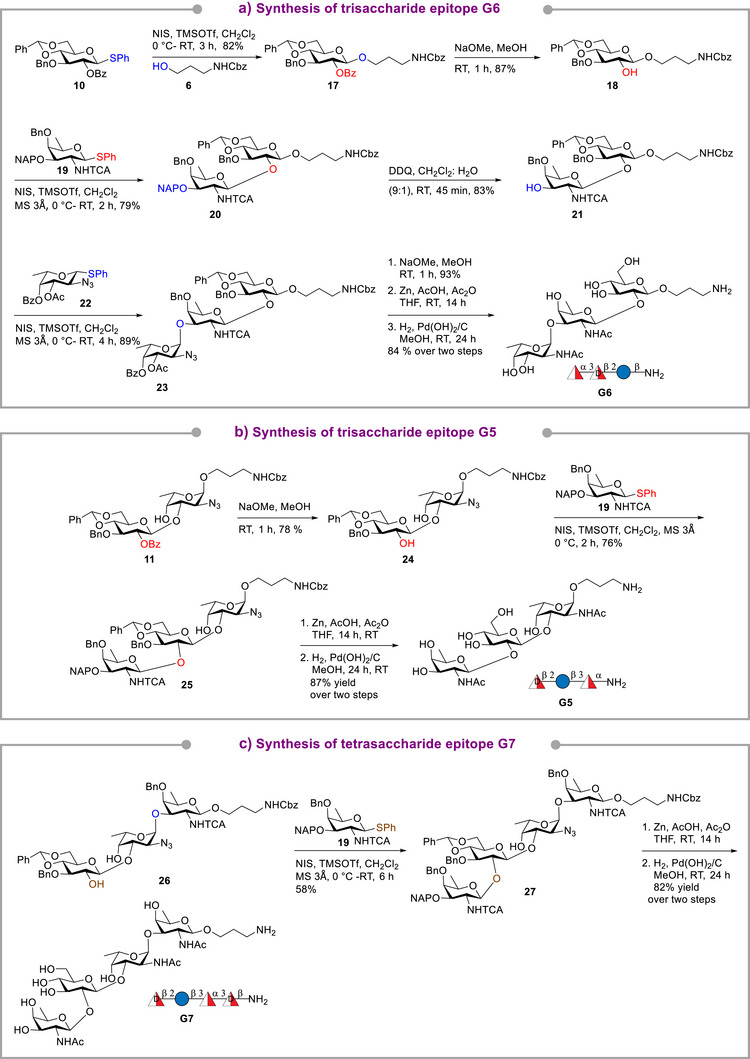 Scheme 3
Scheme 3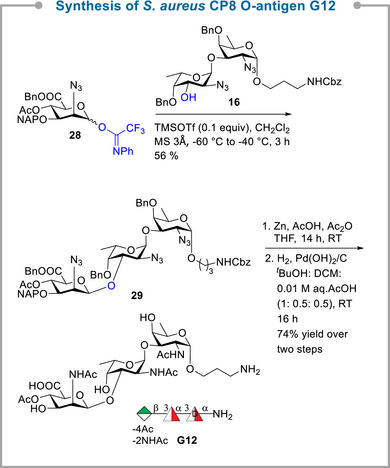 Scheme 4
Scheme 4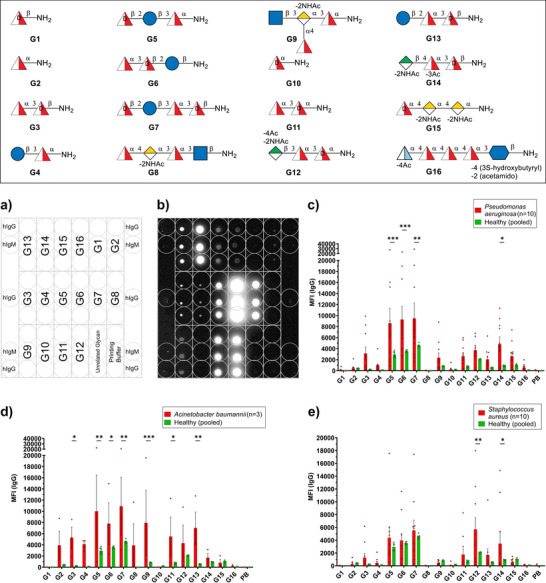 Figure 3
Figure 3| Pathogens | Glycan fragments |
|---|---|
|
| G1–G7 and G13 |
|
| G10–G12 |
|
| G1, G3, and G14 |
|
| G10 and G15 |
|
| G2 and G8 |
|
| G2 and G9 |
|
| G2 and G16 |
- —Science and Education Research Board
- —German Federal Ministry of Research
- —German Research Foundation10.13039/501100001659
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCarbohydrate Chemistry and Synthesis · Antibiotic Resistance in Bacteria · Antimicrobial agents and applications
Introduction
Multidrug‐resistant bacteria are one of the greatest threats to human health today, highlighted by the bacterial‐priority pathogens list issued by the World Health Organization (WHO).^[^ 1, 2, 3 ^]^ The Gram‐negative bacterium Acinetobacter baumannii is resistant to last‐generation antibiotics and has been categorized as a “critical‐priority” pathogen.^[^ 4 ^]^ Similarly, due to their resistance to last‐generation antibiotics, high transmissibility, and mortality rates, especially in healthcare facilities, Pseudomonas aeruginosa and Staphylococcus aureus are classified as “high‐priority” pathogens.^[^ 2, 5 ^]^ Improved diagnostics, prevention, and treatments are needed to control these bacteria.
P. aeruginosa, S. aureus, and A. baumannii are involved in a variety of polymicrobial infections and modulate each other's growth and resistance.^[^ 6, 7, 8, 9, 10, 11 ^]^ These three bacteria belong to a common group of pathogens called ESKAPE pathogens, composed of Enterococcus faecium, S. aureus, Klebsiella pneumoniae, A. baumannii, P. aeruginosa, and Enterobacter spp. They were initially identified as critical multidrug‐resistant (MDR) bacteria with an urgent need for effective treatments. These opportunistic bacteria share traits that allow them to thrive in healthcare environments, including a range of resistance mechanisms rendering them a leading cause of drug‐resistant infections.^[^ 12 ^]^
Glycoconjugate vaccines are a good option to prevent infections with pathogens.^[^ 13, 14, 15, 16 ^]^ A detailed understanding of the postinfection immune response is critical for vaccine development, particularly for the identification of glycan epitopes that may be involved in mediating protective or immunomodulatory effects. Glycan microarrays containing well‐defined, pure synthetic glycans provide a high‐throughput platform for the rapid analysis of serum antiglycan antibodies and the discovery of new cross‐species specific biomarkers.^[^ 17 ^]^ Synthetic access to pure, structurally‐defined bacterial glycotopes containing rare amino‐sugars, equipped with suitable linkers for protein conjugation is essential to enable biomarker discovery. Synthetic epitopes free of biological contaminants are valuable tools for immunological studies. Significant progress has been made in the chemical synthesis of bacterial glycoconjugates containing rare deoxy‐amino sugars.^[^ 18, 19 ^]^ A range of glycoconjugates, including the K. pneumoniae O1 and O2c antigens,^[^ 20 ^]^ Streptococcus pneumoniae capsular polysaccharides (CPS),^[^ 21 ^]^ and Francisella tularensis strain 15^[^ 22 ^]^ were explored using glycan microarrays.^[^ 23 ^]^
P. aeruginosa can cause severe and life‐threatening infections such as pneumonia and bloodstream infections. As an opportunistic pathogen, it poses a serious risk to hospitalized patients.^[^ 24, 25 ^]^ This Gram‐negative bacterium is especially dangerous for immunocompromised individuals and exhibits resistance to most antibiotics, including carbapenems, the few remaining treatment options for multidrug‐resistant infections. Additionally, P. aeruginosa plays a key role in the development of chronic lung infections in patients with cystic fibrosis,^[^ 26 ^]^ non‐cystic fibrosis bronchiectasis (NCFB) and chronic obstructive pulmonary disease (COPD).^[^ 27, 28 ^]^ For decades, vaccines and monoclonal antibodies have been explored for both active and passive immunization against P. aeruginosa. Following some promise in preclinical studies, only one vaccine candidate progressed to clinical trials, and none received approval. Currently, no effective vaccine or therapeutic is available for P. aeruginosa.^[^ 12 ^]^
Another “high‐priority” ESKAPE pathogen, S. aureus is a Gram‐positive bacterium that causes a range of infections, including skin and soft tissue infections as well as device‐related infections.^[^ 29 ^]^ It threatens vulnerable populations such as newborns, surgical patients, and immunocompromised individuals.^[^ 30 ^]^ Methicillin‐resistant S. aureus (MRSA) and vancomycin‐resistant S. aureus (VRSA) present a major public health threat.^[^ 31 ^]^ MRSA is resistant to most β‐lactams, including anti‐staphylococcal penicillins and cephalosporins,^[^ 32 ^]^ and cause approximately 10,600 deaths annually in the USA alone.^[^ 33 ^]^ Among the 12 known S. aureus CPS serotypes, serotypes 5 and 8 account for 85% of clinical isolates, making them attractive targets for vaccine development.^[^ 34 ^]^ A bivalent conjugate vaccine, StaphVAX was developed using CPS types 5 and 8 conjugated to a nontoxic recombinant form of P. aeruginosa exotoxin A and demonstrated protection against S. aureus bacteremia for up to ten months in a phase III clinical trial.^[^ 31, 35 ^]^ Despite the initial success, the vaccine failed to significantly reduce the incidence of invasive S. aureus infections.^[^ 36 ^]^ Vaccine efficacy needs to be improved, potentially by modulating the antigenic construct.^[^ 37 ^]^
A. baumannii, a Gram‐negative coccobacillus^[^ 38, 39 ^]^ can be present on environmental surfaces, is resistant to many disinfectants and spreads within healthcare units.^[^ 38 ^]^ Like other ESKAPE pathogens, it is resistant toward a wide‐range of antibiotics. The CPS of A. baumannii covers the outer membrane and is composed of oligosaccharide units (K units) that serve as key virulence factors protecting the bacteria and helping it evade the host immune response.^[^ 39 ^]^ A. baumannii biofilms significantly reduce the effectiveness of antibiotics and combination therapies.^[^ 40, 41 ^]^ Vaccination offers a strategy to combat multidrug‐resistant strains. Developing a prophylactic vaccine targeting surface polysaccharides is well‐established,^[^ 42 ^]^ but despite extensive research, no effective vaccine has yet been developed for P. aeruginosa, S. aureus, or A. baumannii. These pathogens contain common rare‐amino sugar units, d‐ and l‐N‐acetyl‐fucosamine (FucNAc) (Figure 1). P. aeruginosa O11 and S. aureus (serotypes 5 and 8) exhibit striking structural similarities, sharing a common disaccharide unit that differs only in regio‐ and stereochemistry of the extended linkages. The trisaccharide repeating unit (RU) of S. aureus strain M also contains a d‐FucNAc unit. Likewise, the A. baumannii O5 and S34 RU contain the l‐FucNAc motif. The lipopolysaccharide of Plesiomonas shigelloides strain 302‐73 serotype O1 is comprised of three l‐FucNAc units linked together. These commonalities inspired our exploration into the chemical synthesis of glycan fragments and their analysis using glycan microarrays. Considering the pathogenicity and drug resistance of these bacteria, vaccine development targeting their glycan structures remains a promising strategy, and synthetic glycan fragments offer a powerful tool for probing immune responses to varied epitopes.
Structures of bacterial glycans of different strains of ESKAPE pathogens containing rare d‐FucNAc and l‐FucNAc sugars.
Here, we report the synthesis of a series of glycans (G1–G7 and G10–G12) related to the ESKAPE pathogens: P. aeruginosa O11, S. aureus strains CP5, CP8, and strain M, and A. baumannii S34 and O5 as well as P. shigelloides strain 302‐73 (serotype O1) followed by comparative glycan microarray analyses of G1–G16 (Figure 2 and Table 1). The total syntheses of oligosaccharide RUs from A. baumannii S34 and O5, P. aeruginosa O11, S. aureus CP5, CP8 and strain M, and P. shigelloides serotype O1 including G8, G9, and G13‐ G16 were accomplished previously,^[^ 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53 ^]^ but a systematic microarray‐based comparison of these glycotopes was not performed. Our syntheses focused on the glycan fragments: mono‐(G1, G2, G10), di‐(G3, G4, G11), tri‐(G5, G6, G12), and tetrasaccharide (G7) derivatives. Glycans G8, G9, and G13–G16 were synthesized earlier.^[^ 43, 44, 45, 46, 47 ^]^
Structures of target molecules for microarray investigation.
The trisaccharide RU of P. aeruginosa O11, comprised of →2)‐β‐d‐Glc‐(1→3)‐α‐l‐FucNAc‐(1→3)‐β‐d‐FucNAc‐(1→, a potential vaccine candidate^[^ 54, 55 ^]^ and the RU of S. aureus CP8 →3)‐β‐d‐ManNAcA(4OAc)‐(1→3)‐α‐l‐FucNAc‐(1→3)‐α‐d‐FucNAc‐(1→^[^ 56 ^]^ share a common disaccharide core, differing only in the stereochemistry at their reducing‐end linkages. The syntheses of these glycans pose challenges including rare sugars, 1,2 cis‐linkages including β‐mannosylation and stereoselective linker installation. Additional complexities were encountered in preserving the O‐acetyl substituent at the C4 position of d‐ManNAcA and the incorporation of carboxylates and acetamides in the S. aureus CP8 glycans. The glycan syntheses established a robust platform for accessing diverse glycotopes and enable high‐throughput glycan microarray analyses.
Results and Discussion
The synthesis of glycan epitopes G1–G7 and G10–G12 began with readily available monosaccharides, including d‐mannose, l‐rhamnose, d‐glucose, and α‐methoxy d‐glucose, which were elaborated into FucNAc, Glc, and ManNAcA derivatives. The assembly strategy was designed to enable precise installation of α‐ and β‐glycosidic linkages while maintaining functional groups for subsequent conjugation. The synthesis of three monosaccharide sugar units (G1, G2, and G10) is outlined in Scheme 1. The preparation of intermediate 1 from d‐mannose involved a 16‐step reaction sequence to obtain the desired product in 19% overall yield.^[^ 43 ^]^ Subsequent hydrogenolysis of the protected d‐fucosamine derivative 1 facilitated the global deprotection and simultaneous conversion of NHTCA to acetamide (NHAc) groups, affording target epitope G1 in 98% yield. Further, l‐fucosamine derivative 2 was accessed from l‐rhamnose through an established 11‐step protocol^[^ 43, 45 ^]^ affording the desired product in 21% overall yield. The C2‐azide was then converted to an acetamide group via zinc‐mediated reduction and acetylation in same pot, followed by hydrogenolysis, providing target epitope G2 in 85% yield over two steps. The synthesis of α‐linked d‐fucosamine precursor 3 was accomplished via an established protocol involving a 14‐step reaction sequence, starting from d‐mannose with an overall 26% yield.^[^ 43 ^]^ Oxidative cleavage of 2‐naphthylmethyl (NAP) group in 3 using DDQ afforded 4 in 87% yield which upon subsequent acetylation using acetic anhydride furnished 95% yield of 5. Attempts at the direct glycosylation of highly reactive linker acceptor 6 using donors 3 and 5 and NIS/TfOH or NIS/TMSOTf activation in diethylether as a participating solvent furnished α/β mixtures of d‐fucosamine glycoside. Failure to obtain exclusive α‐selectivity prompted attempts at S* N 2 glycosylations employing glycosyl halide‐mediated in‐situ anomerization. To that end, the NAP ether protecting group was replaced with an electron‐withdrawing acetyl ester to ensure the stability of the bromo intermediate required for stereoselective coupling with 3‐aminopropyl linker 6. This stereoselective glycosyl‐halide mediated S N *2 reaction^[^ 57 ^]^ afforded d‐fucosamine derivative 7 in 62% yield over two steps. Here, TBAI generates a *β *‐glycosyl iodide intermediate via in situ halide exchange, forcing the nucleophile to attack exclusively from the bottom face and thereby delivering exclusively α−glycoside.^[^ 58, 59 ^]^ De‐esterification of 7 and azide to acetamide conversion followed by hydrogenolysis using a solvent mixture (tBuOH:CH_2_Cl_2_:H_2_O in 2:1:1 ratio), furnished G10 in 88% yield.
Synthesis of linker appended monosaccharide glycans G1 (a), G2 (b), and G10 (c).
The syntheses of disaccharide epitopes G3, G4, and G11 started with disaccharide diol 9, glucose 10 and l‐fucosamine 14, respectively.^[^ 43 ^]^ For the synthesis of G3, the azide and NHTCA groups were first converted to acetamide using zinc, acetic acid, and acetic anhydride, followed by hydrogenolysis, to furnish the desired disaccharide epitope in 83% yield. The synthesis of G4, relied on the regioselective union of glucose donor 10 and l‐fucosamine diol acceptor 2 ^[^ 44 ^]^ in the presence of NIS/TMSOTf at 0 °C to afford O3‐linked disaccharide 11 in 66% yield, as a single isomer (Scheme 2). The regioselectivity of disaccharide 11 was confirmed by acetylation of the free C4‐hydroxy group to afford compound S1 (see Supporting Information). A doublet of H4 at δ 5.23 ppm with J = 2.9 Hz in ^1^H NMR spectrum correlating with C4 at δ 68.6 ppm in ^1^H‐^13^C HSQC NMR spectrum, and correlation of ‐CH_3_ proton with H5, and consecutively H5 with H4, and H4 with H3 in the ^1^H − ^1^H COSY spectrum along with HMBC correlations confirmed the presence of the acetyl group at the C‐4 position and eventually the glycosidic bond at C‐3 of S1. The transformation of the azide into an acetamido group using zinc‐mediated reduction followed by acetylation furnished 12 in 78% yield. Finally, debenzoylation of 12 using NaOMe in MeOH gave diol 13 in 92% yield, before hydrogenolysis furnished disaccharide epitope G4 in 97% yield. Disaccharide G11 was prepared by coupling l‐fucosamine donor 14 ^[^ 44 ^]^ with d‐fucosamine acceptor 8 and NIS/TMSOTf using solvent participation, (CH_2_Cl_2_:Et_2_O; 1:3) at −78 °C furnished 1,2‐cis linked disaccharide 15 as a sole isomer in 82% yield. Oxidative cleavage of the NAP ether afforded disaccharide 16 in 78% yield. Azide to acetamide conversion followed by hydrogenolysis afforded disaccharide epitope G11 in 79% yield.
Synthesis of the disaccharide fragments from P. aeruginosa O11 and S. aureus CP5 and CP8. Synthesis of G3 (a), G4 (b) and G11 (c).
The synthesis of tri‐ (G5 and G6) and tetrasaccharide (G7) epitopes of P. aeruginosa O11 (Scheme 3) started with coupling of the aminopropyl linker with glucose 10 followed by debenzoylation to furnish 87% yield of acceptor 18. Utilizing NHTCA neighboring group participation in d‐fucosamine19 ^[^ 43 ^]^ ensured selective glycosylation of 18 to give β‐linked disaccharide 20 in 79% yield. Oxidative cleavage of the 2‐naphthylmethyl group in 20 using DDQ in CH_2_Cl_2_:H_2_O (9:1) afforded disaccharide acceptor 21 in 83% yield. Utilizing long‐range participation via C4‐OBz in donor 22, α‐stereoselective glycosylation of disaccharide acceptor 21 yielded exclusively fully functionalized trisaccharide 23 in 89% yield. De‐esterification of trisaccharide 23, afforded 93% of the corresponding diol, followed by conversion of the azide and NHTCA groups to acetamido and hydrogenolysis using Pd(OH)2/C in methanol yielded trisaccharide glycotope G6 in 84% yield. (Scheme 3a). For the synthesis of G5, debenzoylation of 11 gave disaccharide acceptor 24 in 78% yield. Regioselective coupling of the equatorial C2‐OH in 24 with donor 19 in the presence of NIS/TMSOTf afforded the 1,2‐trans O2 coupled trisaccharide 25 in 76% yield. Azide and NHTCA were converted to NHAc, followed by hydrogenolysis to furnish trisaccharide epitope G5 in 87% yield (Scheme 3b). The synthesis of tetrasaccharide G7 started with a regioselective 1,2‐trans coupling of trisaccharide diol 26 with donor 19 at 0 °C to afford tetrasaccharide 27 in 58% yield. Treatment of the protected tetrasaccharide with Zn, AcOH, Ac_2_O and hydrogenolysis delivered desired epitope G7 in 82% yield (Scheme 3c). The position of glycosidic bonds in oligosaccharides such as G5 and G7 were ascertained by extensive 2D NMR experiments (see Supporting Information).
Synthesis of trisaccharides G6 (a), G5 (b) and tetrasaccharide G7 (c) of P. aeruginosa O11.
The synthesis of the S. aureus type 8 trisaccharide epitope G12 commenced with the coupling of trifluoroacetimidate donor 28 ^[^ 44 ^]^ and disaccharide acceptor 16. Earlier, we reported the synthesis of a trisaccharide RU of CP8 bearing a PMP group at the reducing end, wherein the installation of a similar β‐mannosidic linkage was achieved using excess glycosyl donor at low temperature. Since a linker is necessary for microarray studies, glycosylation of the linker‐appended disaccharide acceptor 16 (Scheme 2c) was attempted at −60 °C using TfOH (0.5 equiv) as a promoter. The reaction proceeded in low yield with a lots of unreacted acceptor 16 remaining. Conducting the reaction −60 °C using TMSOTf (0.1 equiv) while slowly increasing the temperature to −40 °C over 3 h, gave 56% of trisaccharide 29 (Scheme 4). This reaction is believed to proceed via a half‐chair oxocarbenium intermediate in ^3^ H 4 confirmation with the benzylic ester participating to enforce β‐selectivity.^[^ 60, 61, 62 ^]^ β‐Stereoselectivity was confirmed by J CH coupling experiments (J C1−H1 = 162.2 Hz) (see Supporting Information). Using limited amounts of promoter (0.1 equiv) and excess donor (four equivalents) proved crucial for achieving the desired β‐mannosidic linkage, as excess promoter led to TMS‐protection of the hydroxyl groups. Subsequent reduction of the azides in fully protected trisaccharide 29 to amines followed by acetylation furnished the corresponding NHAc intermediate. Surprisingly, the acetate group at C‐4 was found to be highly labile, migrating and/or cleaving under certain conditions (PhSH/Et_3_N or hydrogenolysis in methanol). We believe that the free amino group in the linker may promote acetate migration or partial deacetylation, particularly during prolonged hydrogenation in methanol. Hydrogenolysis in a solvent mixture of tBuOH:CH_2_Cl_2_:0.01 M aq. AcOH (1:0.5:0.5) and Pearlman's catalyst for 16 h afforded the final S. aureus CP8 epitope G12 in 74% yield over two steps.
Synthesis of S. aureus CP8 trisaccharide RU G12.
The synthetic glycans were immobilized in triplicates on N‐hydroxy succinimide (NHS)‐activated glass slides to detect binding of IgG antibodies in sera of patients infected with P. aeruginosa, A. baumannii, or S. aureus (Figure 3). Patients infected with P. aeruginosa showed a higher IgG antibody level against the P. aeruginosa strain O11‐related G5, G6, and G7 compared to healthy individuals. IgG antibodies against the S. aureus type 5 related G14 were detected in addition. IgG antibodies directed toward A. baumannii strain O5‐related glycan G9 were present in the sera of patients infected with A. baumannii. Furthermore, P. aeruginosa strain O11‐related G3, G5, G6, G7, and G13 and S. aureus CP8‐related G11 were epitopes for IgG antibody binding. All these structures contain both l‐FucNAc and d‐FucNAc monosaccharides. S. aureus infected patients showed IgG antibodies against the S. aureus CP8‐related G12 and S. aureus type 5‐related G14 and no binding toward G11, leading to the conclusions that the trisaccharide is the minimal epitope and the d‐FucNAc glycosidic linkage with linker is of no significance. Since current diagnostic tools do not differentiate between strains of bacterial infections, not all patients showed antibodies toward the bacteria‐related glycans. IgG antibodies directed against G5, G6, G7, and G12 were detected in healthy patients, potentially a result of previous infections with related bacterial species. The microarray results show that although the trisaccharide is the minimal epitope, the sequence and stereochemistry of the d‐Glc, l‐FucNAc, and d‐FucNAc residues have an influence on antibody binding. Glycans G5, G6, and G13 contain the same monosaccharides but differ in connectivity, resulting in different immunogenicity. These subtle topological differences explain why G6, and not the closely related G13, emerged as the lead structure together with G12 and G14.
Glycan microarray analysis of sera derived from patients infected with P. aeruginosa, A. baumannii or S. aureus. Glycan microarray printing pattern (a) and exemplary binding pattern of one human serum to immobilized synthetic glycans (b). Mean fluorescence intensity (MFI) of IgG antibody binding to synthetic glycans in patients infected with P. aeruginosa (c), A. baumannii (d), or S. aureus (e). A serum dilution of 1:100 was used. Values represent mean ± SEM. Differences were tested for significance to pooled sera of healthy individuals using multiple Two‐Way ANOVA test with () p < 0.0002, () p < 0.002 and () p < 0.033.
Conclusion
We synthesized glycans that structurally mimic O‐antigens and CPS belonging to three pathogens from the ESKAPE family, P. aeruginosa O11, S. aureus (CP5, CP8, and strain M), A. baumannii (S34 and O5), as well as P. shigelloides O1. These glycans share rare deoxy amino sugars, notably d‐fucosamine and l‐fucosamine. Our synthetic route to the target glycans relied on regio‐ and stereoselective glycosylations to avoid extensive protection/deprotection sequences. The formation of the requisite 1,2‐cis glycosidic linkages was controlled by leveraging both solvent and long‐range participation effects. The terminal aminopropyl linker was installed onto the d‐FucNAc moiety, via in situ anomerization protocol involving a bromo intermediate, to furnish exclusively the desired α‐linked product. A direct β‐mannosylation employing a N‐phenyl‐2,2,2‐trifluoroacetimidoyl chloride donor with a rare disaccharide acceptor facilitated expedient access to a conjugation‐ready trisaccharide repeating unit of S. aureus CP8, a promising vaccine candidate.
A total of sixteen glycans were evaluated in order to get insights into the antibody production after the infection with P. aeruginosa, S. aureus or A. baumannii to define key immunogenic epitopes. Glycan microarray analysis of sera derived from patients after infection with P. aeruginosa, S. aureus or A. baumannii showed significantly higher antibody levels against the trisaccharides G14, G6, and G12 compared to the level in healthy humans. Similar glycan composition across different strains of bacteria renders synthetic trisaccharides G14, G6, and G12 a promising option for cross‐protective immunization against all three bacteria.
Future investigations will focus on detailed antigen–antibody binding studies to uncover molecular recognition patterns, along with animal immunization experiments to assess protective efficacy. This synthetic platform broadens access to rare‐sugar‐containing bacterial glycans for next‐generation carbohydrate‐based vaccines and diagnostics tools.
Conflict of Interests
The authors declare no conflict of interest.
Supporting information
Supporting Information
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Antimicrobial Resistance Collaborators (ARC) , Lancet 2022, 399, 629–655, 10.1016/S 0140-6736(21)02724-0.35065702 PMC 8841637 · doi ↗ · pubmed ↗
- 2T. Jesudason , Lanc. Micr. 2024, 5, 100940, 10.1016/j.lanmic.2024.07.003.39079540 · doi ↗ · pubmed ↗
- 3Hatim Sati , Elena Carrara , Alessia Savoldi , Paul Hansen , Jacopo Garlasco , Enrica Campagnaro , Simone Boccia , Juan Antonio Castillo‐Polo , Eugenia Magrini , Pilar Garcia‐Vello , Eve Wool , Valeria Gigante , Erin Duffy , Alessandro Cassini , Benedikt Huttner , Pilar Ramon Pardo , Mohsen Naghavi , Fuad Mirzayev , Matteo Zignol , Alexandra Cameron , Evelina Tacconelli , the WHO Bacterial Priority Pathogens List Advisory Group, WHO, WHO Bacterial Priority Pathogens List, 202 · doi ↗ · pubmed ↗
- 4Y. Jiang , Y. Ding , Y. Wei , C. Jian , J. Liu , Z. Zeng , Front. Microbiol. 2022, 13, 1045206, 10.3389/fmicb.2022.1045206.36439795 PMC 9684325 · doi ↗ · pubmed ↗
- 5A. S. Lee , B. D. Huttner , G. Catho , S. Harbarth , Infect. Dis. Clin. North Am. 2021, 35, 931–952, 10.1016/j.idc.2021.07.001.34752226 · doi ↗ · pubmed ↗
- 6N. M. Smith , A. Ang , F. Tan , K. Macias , S. James , J. Sidhu , J. R. Lenhard , Antimicrob. Agents Chemother. 2021, 65, 10–1128, 10.1128/aac.02414-20.PMC 809744733495215 · doi ↗ · pubmed ↗
- 7L. Radlinski , S. E. Rowe , L. B. Kartchner , R. Maile , B. A. Cairns , N. P. Vitko , C. J. Gode , A. M. Lachiewicz , M. C. Wolfgang , B. P. Conlon , P Lo S Biol. 2017, 15, e 2003981, 10.1371/journal.pbio.2003981.29176757 PMC 5720819 · doi ↗ · pubmed ↗
- 8I. Pastar , A. G. Nusbaum , J. Gil , S. B. Chen , J. Patel , J. Valdes , O. Stojadinovic , L. R. Plano , M. Tomic‐Canic , S. C. Davis , P Lo S One 2013, 8, e 56846, 10.1371/journal.pone.0056846.23451098 PMC 3579943 · doi ↗ · pubmed ↗