A Riboflavin‐Derived Flavinium Salt Mediates Chemoselective Methylation Reactions
Tim Langschwager, Ekrem Suylu, Julian Zuber, Golo Storch

TL;DR
A new methylation method using a flavin mediator allows selective and safe methyl group addition to complex molecules.
Contribution
A recyclable flavin-mediated methylation strategy with high chemoselectivity and applicability to biologically relevant substrates.
Findings
Flavin mediator is easily recovered and reused after methylation reactions.
The method enables chemoselective methylation of complex molecules like venetoclax and nevirapine.
Trideuteromethylation and nucleobase modification are promising applications of the method.
Abstract
Selective methylation is among the most relevant transformations in synthetic chemistry and the discovery of new drug molecules. A methyl group is typically installed using strong electrophiles such as methyl iodide or dimethyl sulfate, which are associated with safety hazards and limited chemoselectivity. A promising strategy for circumventing these limitations relies on splitting the methylation into a two‐step procedure under mediator control. However, such reactions, including the Mukaiyama redox condensation, currently lack applicability since the mediator is lost as organic waste, resulting in a low atom economy. We have developed a flavin‐mediated methylation strategy with easily accessible methyl diphenylphosphinite (Ph2POMe) as the source of the methyl group. The flavin mediator is easily recovered by a simple acidic treatment followed by oxidation with air. Detailed NMR…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
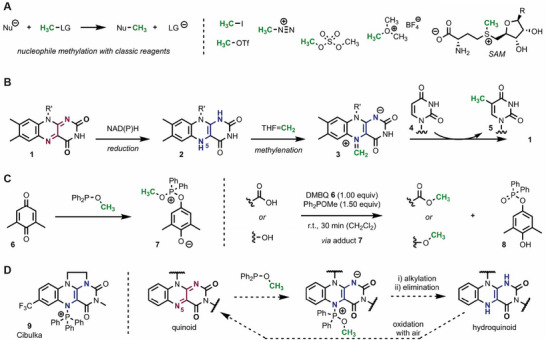 FIGURE 1
FIGURE 1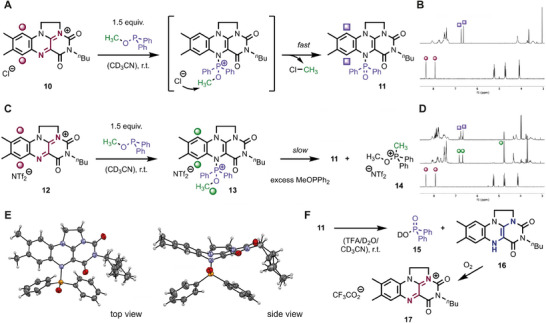 FIGURE 2
FIGURE 2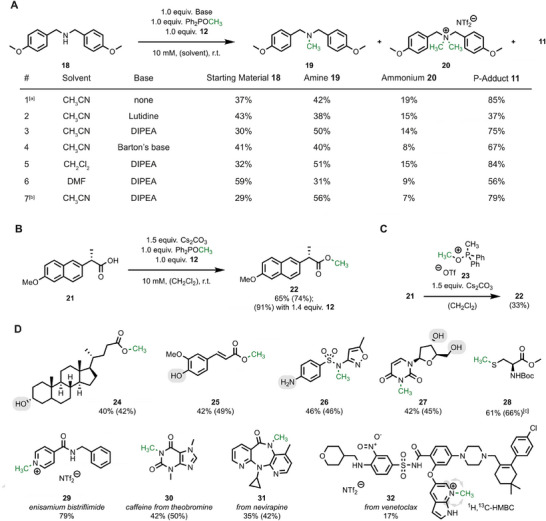 FIGURE 3
FIGURE 3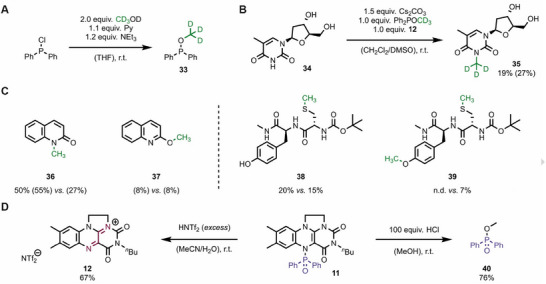 FIGURE 4
FIGURE 4- —Bayerische Akademie der Wissenschaften10.13039/501100007306
- —Fonds der Chemischen Industrie10.13039/100018992
- —Deutsche Forschungsgemeinschaft10.13039/501100001659
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsClick Chemistry and Applications · Catalytic C–H Functionalization Methods · Fluorine in Organic Chemistry
Introduction
1
Alkylation reactions are broadly applied transformations in organic chemistry. Methylation is a prominent case amongst this reaction class with widespread applications in pharmaceutical chemistry. Known as the magic methyl effect, interest in this particular modification stems from the often substantial increase in biological activity by seemingly simple addition of a CH_3_ group [1, 2]. Several methodologies for methylation have been developed, including reductive alkylation [3], transition‐metal protocols [4, 5], and photochemical approaches [6]. While a methyl group can also be installed by using a methyl nucleophile such as methyl lithium, electrophilic methylation is most broadly applicable since heteroatoms and carbanions are typical positions for desired functionalization. However, these reactions require strong electrophiles, which are prone to unselective reactions and pose significant health risks (Figure 1A). Therefore, we took the opportunity to explore orthogonal methylation reactions that circumvent these limitations.
Overview of selected methylation strategies. (A) Classic methylation reagents in synthesis and in biology. Nu = nucleophile; LG = leaving group; R = 6‐aminopurinyl. (B) Methylation with flavin‐dependent enzymes via a two‐step mechanism. R’ = Ribityl‐adenosine diphosphate. (C) The Mukaiyama redox condensation. (D) Our conceptual idea of a flavin‐mediated methylation with regeneration of the mediator.
The archetypal methylation reaction in nature—alkylation with S‐adenosyl methionine (SAM) [7, 8]—also follows the paradigm outlined above, since a strong sulfonium electrophile serves as the active species. In contrast, flavin‐dependent methyltransferases enable alkylation by an intriguing combination of two independent reaction steps (Figure 1B). The quinoid flavin cofactor (1) is initially reduced to the hydroquinone 2, and 5,10‐methylenetetrahydrofolate (THF = CH_2_) transfers its methylene unit to the flavin N5 position, which leads to electrophilic iminium adduct 3 [9, 10, 11]. In the case of thymidylate synthase ThyX, the enzymatic substrate deoxyuridine monophosphate (dUMP, 4) then attacks with its nucleophilic position, and the methylated analog 5 is formed together with quinoid flavin 1 [12].
In organic synthesis, the Mukaiyama redox condensation also segments the alkylation reaction into two separate steps [13, 14]. The transformation relies on a phosphinite reagent such as methyl diphenylphosphinite (Ph_2_POMe), which undergoes nucleophilic attack on the electrophilic 2,6‐dimethylbenzoquinone (DMBQ, 6) and generates the active alkylating agent 7 (Figure 1C) [15, 16]. The strategy allows turning two relatively benign reagents into a reactive species in situ, and subsequent alkylation typically of oxygen heteroatoms. While elegant from a conceptual standpoint, the benzoquinone is not regenerated and ultimately collected as organic waste in the form of hydroquinone 8. Similar to the Mukaiyama redox condensation, Wang et al. broadened the scope of this reaction by exchanging DMBQ with ethyl acrylate, which, however, still led to the stoichiometric production of phosphinoxide byproducts [17].
Building on the general similarity between benzoquinones and flavins, we hypothesized that the latter might also react with phosphinites, leading to active methylating species. While several reversible adduct formations of flavins are known in enzymes and model systems [18, 19, 20, 21], only the reversible addition of phosphines to modified synthetic flavins was reported by Cibulka [22, 23]. Their results document that triphenylphosphine readily adds to cationic flavinium salts [24], leading to N5 adduct 9. If possible with phosphinites, this would ultimately lead to an electrophilic species similar to the one formed in the Mukaiyama redox condensation (Figure 1D). Regeneration of the initial quinoid flavin would then require elimination and oxidation of the hydroquinoid form—two steps that are well‐explored and feasible with organic flavin cofactors.
Results and Discussion
2
Our initial interest was centered around the so far unexplored reactivity of flavinium salt 10 with phosphinites. The flavinium salt was prepared by a modified literature procedure in only four steps from (–)‐riboflavin (see Supporting Information for details) [24, 25]. This compound has improved solubility properties and is accessible without a single chromatographic purification step. In a promising first experiment, we directly observed a clean reaction with methyl diphenylphosphinite in deuterated acetonitrile solution (Figure 2A). Analysis of the NMR spectra (Figure 2B) revealed that the sole flavin‐containing product is P‐adduct 11, which was identified in combination with chloromethane in solution (^1^H NMR: δ = 3.03 ppm) [26]. The latter gaseous compound was presumably observed in solution since these experiments were conducted in Teflon screw‐cap NMR tubes. While unproductive for the desired substrate methylation, this result already confirmed that the intermediate is an activated methylation agent that transfers the methyl group to the moderately nucleophilic chloride anion. A logical step toward circumventing this undesired anion methylation was to use flavinium bistriflimide 12 in the analogous reaction (Figure 2C). This compound was prepared in a single step by treating chloride 10 with AgNTf_2_ in 88% yield (see Supporting Information for details). Indeed, these conditions allowed characterization of the immediate adduct 13 in solution by proton NMR spectroscopy and high‐resolution ESI‐MS (Figure 2D and Supporting Information). A ^3^ J H‐P coupling constant of 12.0 Hz was observed between the protons at the methyl group and the phosphorous atom, indicative of an intact C─O bond. No reaction with the bistriflimide anion was observed. However, prolonged reaction times led to methylation of the excess methyl diphenylphosphinite and generation of salt 14. Having confirmed the formation and reactivity of the immediate adduct 13, we next focused on the neutral adduct 11. While we initially suspected the compound to be sensitive toward moisture and air due to its structural relation to hydroquinoid flavins, it turned out to be relatively stable and could even be isolated as a faint‐orange solid. Single‐crystal analysis (Figure 2E) confirmed the connectivity and the new P─N bond at the flavin N5 position. The side view clearly indicates the bent flavin geometry, indicative of the reduced state. To the best of our knowledge, this is the first single‐crystal structure of a flavin adduct with heteroatom substitution at the N5 position. While stable under neutral conditions, we found that subjecting adduct 11 to trifluoroacetic acid (TFA) in deuterated acetonitrile and deuterated water leads to conversion. We observed hydrolysis of the adduct concomitant with an oxidation of the formed reduced flavin by air (Figure 2F). These results already indicate that the flavinium salt 12 might be recyclable from adduct 11.
Reaction of flavinium salts with methyl diphenylphosphinite. (A) Reaction scheme in deuterated acetonitrile. (B) Comparison of the proton NMR spectra. (C) The analogous reaction with the NTf2 salt. (D) NMR spectra of formation and slow conversion of the active electrophile 13. (E) Molecular structure of adduct 11 with a disordered butyl group obtained by single‐crystal X‐ray diffraction. (F) Reactivity of adduct 11 under acidic conditions.
We then explored the activity of phosphinite adduct 13 in alkylation reactions with organic substrates. The secondary amine 18 served as an informative model substrate since several challenges arise here with alkylation reactions, including overalkylation to the ammonium salt. The flavinium bistriflimide 12 was chosen, activated with commercially available Ph_2_POCH_3_, and combined in equimolar ratio with amine 18 (Figure 3A; for a full screening of conditions, see Supporting Information). Indeed, we observed substrate methylation to tertiary amine 19 as the major product, but overalkylation to ammonium salt 20 occurred with considerable amounts (Entry 1). Since the net reaction includes formation of an HNTf_2_ equivalent, we hypothesized that the slightly more basic secondary amine substrate might be protonated predominantly, hindering its activity as a nucleophile. Following this rationale, methylation of the tertiary amine 19 is preferred. A quick screen of several bases (Entries 2 – 4) confirmed that selectivity of the methylation was improved under these conditions, with DIPEA being a good compromise of reactivity and selectivity. Dichloromethane as solvent performed equally well, while DMF provided decreased levels of conversion (Entries 5 and 6). Slow addition of the activated flavin adduct over a period of 30 min turned out to be beneficial and led to 56% yield of the tertiary amine, while only 7% of the ammonium salt were formed (Entry 7). The flavin‐mediated methylation protocol is not limited to amine nucleophiles, and slight adaptations in solvent and base led to a straightforward method for methylation of (S)‐naproxen 21 to the corresponding methyl ester 22 (Figure 3B). An even higher yield of 91% was observed with a small excess of flavin 12 (0.4 equiv.) and phosphinite. We also studied the possibility that the methylated phosphinite 14 might serve as an additional methylating agent (Figure 3C). While competent under isolated conditions (tested with triflate 23), the contribution of this pathway in the flavin‐mediated reaction is marginal since only one equivalent of Ph_2_POMe is used, and 79% of flavin adduct 11 was observed (c.f., Figure 3A Entry 7).
Methylation reaction with flavinium salt 12 and Ph2POMe. (A) Secondary amine substrate and reaction screening. Percentages refer to yields determined by NMR spectroscopy with an internal standard. (B) Esterification of (S)‐naproxen using the flavin methodology. (C) Probing for the alkylation ability of salt 23 in the esterification reaction. (D) Scope of the selective methylation reaction. Yields in parentheses refer to NMR experiments with an internal standard, all other values correspond to isolated material. [a] Reaction set up at −30 °C and warmed up to room temperature (r.t.). [b] Adduct 13 was added by syringe pump over a period of 30 min. Yield of isolated material of amine 19 is 46%. [c]: Here, 1.5 equiv. phosphinite were used.
Having established an easily adaptable methodology, we investigated the substrate scope with increasing levels of complexity (Figure 3D). Carboxylic acids were smoothly methylated in the presence of alcohols and phenols, as exemplified by the esters of lithocholic acid (24) and ferulic acid (25). Phenolates and carboxylates usually possess similar nucleophilicity (N = 16.45 for benzoate in MeCN and N = 18.53 for phenolate in MeCN) [27, 28]. Accordingly, product mixtures were obtained from ferulic acid with Ph_2_POCH_3_ and ethyl acrylate (see Supporting Information for details) [17]. Selective methylation was observed for the antibiotic drug sulfamethoxazole, where the aniline did not react, and sole methylation of the sulfonamide was obtained (26). Similarly, methylation of the imide in desoxyuridine (27) proceeded without any concurrent methylation of the oxygen nucleophiles. The thiol position in a protected cysteine (28) was also confirmed as a suitable nucleophile for methylation. Our mild methylation protocol also provides access to selective methylation in complex molecules. Enisamium bistriflimide (29) was prepared by methylation of the pyridine nitrogen position in 79% yield, and caffeine (30) resulted in 50% yield from methylation of theobromine. Despite the presence of four competing nucleophilic positions, methylation of the HIV drug nevirapine occurred with high site‐selectivity and led to analog 31 in a yield of 42%. Venetoclax, a drug molecule for treating leukemia, was also subjected to our flavin‐mediated methylation protocol, which led to the isolation of derivative 32. These reactions clearly highlight that the combination of flavinium salts and phosphinites is beneficial for selective methylation under mild conditions.
The broad applicability of our flavin‐mediated methylation methodology was then explored in detail. Trideuteromethylation was identified as a target reaction given its relevance in drug discovery [29, 30]. Several trideuteromethyl‐containing drug candidates are currently being developed or are approved by the FDA [31], including the tyrosine kinase 2 inhibitor deucravacitinib [32]. As an advantage of our strategy, the required trideuteromethyl diphenylphosphinite 33 is easily prepared from chlorodiphenylphosphine and deuterated methanol in a single step (Figure 4A) [17]. With this reagent in hand, the selective trideuteromethylation of thymidine 34 (Figure 4B) led to the trideutero‐analogue of 3‐meT 35, which is recognized by the fat mass and obesity‐associated (FTO) gene [33]. Inspired by the observed high selectivity of one specific methylation site throughout our study, we also probed difficult selectivity challenges [34]. While classic methylation agents such as methyl triflate only moderately distinguish (3.4 [36]: 1 [37]) between N‐ and O‐alkylation of quinolone, the reaction was both more efficient and more selective (6.9 [36]: 1 [37]) with our flavin‐mediated method (Figure 4C). The observation that phenolic positions are not alkylated (c.f., Figure 3D) encouraged us to test dipeptides containing tyrosine and cysteine. With methyl triflate, we observed cysteine methylation (38) and overalkylation at both positions (39). In contrast, the flavin‐mediated strategy did not lead to overalkylation (see Supporting Information for details). Besides selectivity, the recyclability of the flavin mediator was identified as a key prerequisite for an efficient protocol. Building on our initial observation that adduct 11 is unstable under acidic conditions, we tested the addition of bistriflimidic acid (HNTf_2_) for selective hydrolysis (Figure 4D). Indeed, flavinium salt 12 was isolated in 67% under these conditions, which implies that no ion exchange is necessary before application in the next reaction. In methanolic solution, acidic treatment leads to flavin recovery and the corresponding methyl diphenylphosphinate 40, a useful compound inter alia as a sensitizer [35].
Application of the flavin‐mediated methylation protocol. (A) Preparation of trideuterated phosphinite 33. (B) Trideuteromethylation of thymidine 34. (C) Comparative reactivity of the flavin methylation protocol (left entries) versus methyl triflate (right entries). Conditions: 1.0 equiv. methyl triflate. For conditions of the flavin‐mediated reaction, see Figure 3 and Supporting Information. Yields in parentheses were determined by NMR spectroscopy relative to an internal standard. All other values refer to isolated material. (D) Recycling of the flavinium mediator under acidic reaction conditions.
Conclusion
3
Our study shows that flavinium salts derived from naturally occurring (–)‐riboflavin are competent mediators in redox condensation methylations with organic substrates. We observed high chemoselectivity in several cases, typically avoiding any reaction at the alcohol and phenol positions. These characteristics are expected to serve as beneficial properties for achieving selective functionalization of complex molecules, including biologically active compounds. In this context, the facile incorporation of trideuteromethyl groups likely adds to the synthetic usefulness. Contrary to several existing strategies, the simple recovery of the flavin mediator through acidic workup and oxidation by air also adds to the applicability of our protocol. In addition to methylation, we envision the flavin‐mediated strategy to be equally effective in other group transfer reactions if different phosphinites are applied. With other phosphorous nucleophiles, we expect similar flavin‐mediated reactions to unlock completely new transformations.
Conflicts of Interest
The authors declare no conflict of interest.
Supporting information
The data that support the findings of this study are available in the supplementary material of this article. Deposition number 2496823 (for 11) contains the supplementary crystallographic data for this paper. This data is provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service. The authors have cited additional references within the Supporting Information [36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59].
Supporting file 2: chem70586‐sup‐0002‐DataFile‐ee.zip
Supporting file 3: chem70586‐sup‐0003‐DataFile‐ff.zip
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1H. Schönherr and T. Cernak , “Profound Methyl Effects in Drug Discovery and a Call for New C–H Methylation Reactions” Angewandte Chemie International Edition 52 (2013): 12256–12267, 10.1002/anie.201303207.24151256 · doi ↗ · pubmed ↗
- 2E. J. Barreiro , A. E. Kümmerle , and C. A. M. Fraga , “The Methylation Effect in Medicinal Chemistry,” Chemical Reviews 111 (2011): 5215–5246, 10.1021/cr 200060 g.21631125 · doi ↗ · pubmed ↗
- 3A. A. Boezio , J. Pytkowicz , A. Côté , and A. B. Charette , “Asymmetric, Catalytic Synthesis of α‐Chiral Amines Using a Novel Bis(phosphine) Monoxide Chiral Ligand,” Journal of the American Chemical Society 125 (2003): 14260–14261, 10.1021/ja 038291+.14624558 · doi ↗ · pubmed ↗
- 4T. T. Dang , B. Ramalingam , and A. M. Seayad , “Efficient Ruthenium‐Catalyzed N‐Methylation of Amines Using Methanol,” ACS Catalysis 5 (2015): 4082–4088, 10.1021/acscatal.5b 00606. · doi ↗
- 5M. Giedyk , K. Goliszewskaab , and D. Gryko , “Vitamin B 12 Catalysed Reactions” Chemical Society Reviews 44 (2015): 3391–3404, 10.1039/C 5CS 00165 J.25945462 · doi ↗ · pubmed ↗
- 6V. N. Tsarev , Y. Morioka , J. Caner , et al., “N‐Methylation of Amines With Methanol at Room Temperature,” Organic Letters 17 (2015): 2530–2533, 10.1021/acs.orglett.5b 01063.25915546 · doi ↗ · pubmed ↗
- 7M. R. Bennett , S. A. Shepherd , V. A. Cronin , and J. Micklefield , “Recent Advances in Methyltransferase Biocatalysis,” Current Opinion in Chemical Biology 37 (2017): 97–106, 10.1016/j.cbpa.2017.01.020.28259085 · doi ↗ · pubmed ↗
- 8E. Abdelraheem , B. Thair , R. F. Varela , et al., “Methyltransferases: Functions and Applications” Chem Bio Chem 23 (2022): e 202200212, 10.1002/cbic.202200212.35691829 PMC 9539859 · doi ↗ · pubmed ↗