Photoinduced, Chemoselective γ‐Alkylation of 2‐Silyloxyfurans With α‐Bromoketones: A Rapid Entry to Chiral ε‐Keto‐γ‐Butenolides
Debora Guazzetti, Luca Aimi, Enrico Marcantonio, Giovanni Maria Siciliano, Kelly Bugatti, Sara Dobani, Andrea Sartori, Lucia Battistini, Franca Zanardi, Claudio Curti

TL;DR
This paper introduces a new one-step method to create chiral ε-ketobutenolides using light, which can be used to build complex molecules.
Contribution
A novel photoinduced γ-alkylation method for synthesizing ε-ketobutenolides in high yields.
Findings
The γ-alkylation of 2-silyloxyfurans with α-bromoketones was achieved using blue light.
The method provides access to chiral ε-ketobutenolides in one step and high yields.
The scaffolds were used to create new heterobicyclic derivatives like phenyltetrahydrofurofuran and tetrahydrofuro-pyridazinone.
Abstract
γ‐Butenolides are widespread structural motifs found in many natural and unnatural products which display an impressive range of biological activities. Among them, ε‐keto‐γ‐butenolides represent underestimated butenolide frameworks, which could serve as valuable platforms to build complex structures, for example, heterobicyclic derivatives. Quite unexpectedly, despite the apparent simplicity of their structures, efficient synthetic methodologies enabling the construction of chiral, ε‐keto‐γ‐butenolide architectures are quite underdeveloped. In this context, herein we present a novel, photoinduced regio‐ and chemoselective γ‐alkylation of 2‐silyloxyfurans with 2‐bromoketones providing a practical access to ε‐ketobutenolide scaffolds in racemic format, in one single step and high yields. The usefulness of these products as starting materials to build chiral, fused‐heterobicycle lactone…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 SCHEME 1
SCHEME 1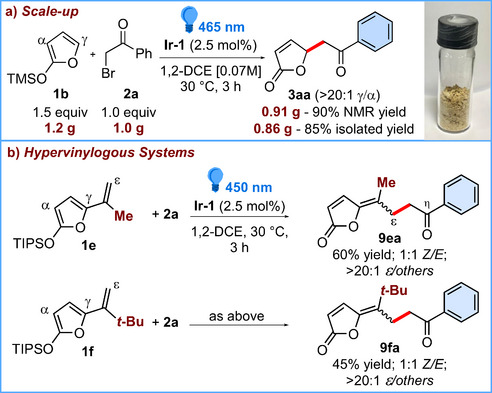 SCHEME 2
SCHEME 2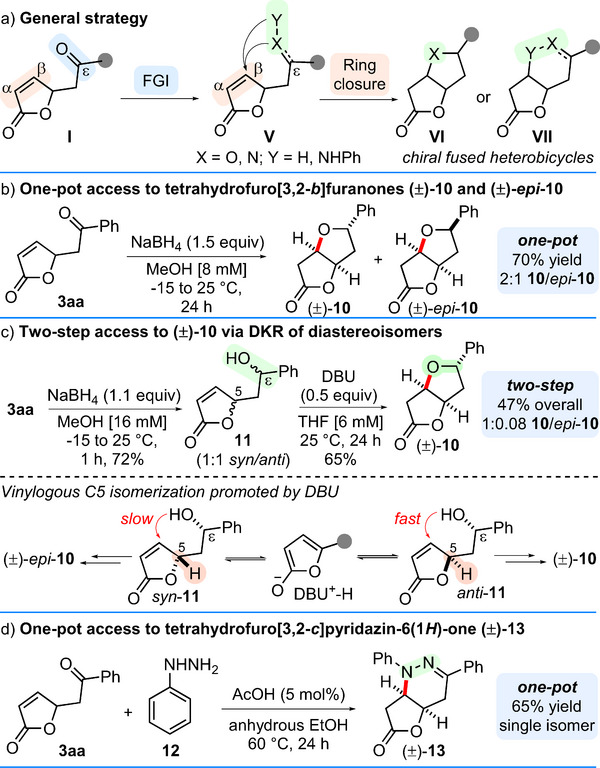 SCHEME 3
SCHEME 3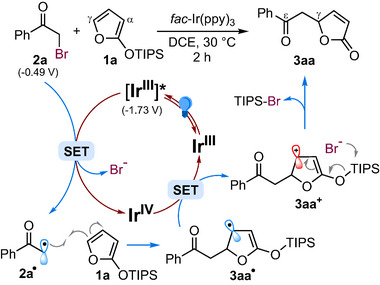 SCHEME 4
SCHEME 4|
| |||
| Entry |
Deviations from the initial conditions |
Yield [%]
|
|
| 1 | None | 35 | 1:0.17 |
| 2 |
| 9 | n.d. |
| 3 |
| 20 | 1: 0.08 |
| 4 | 1,2‐DCE | 42 | 1:0.10 |
| 5 | MeCN |
| |
| 6 | 1,2‐DCE, | 45 | 1:0.10 |
| 7 |
As entry 6 without K2CO3 | 50 | 1:0.06 |
| 8 |
As entry 7 with 1.5:1 ( | 90 | 1:0.03 |
| 9 | As entry 8, for 3 h | 93 | 1:0.03 |
| 10 |
As entry 9, with | 88 | 1:0.01 |
| 11 | No photocatalyst |
| |
| 12 | No light |
| |
|
| |||
|
|
- —H2020 European Research Council10.13039/100010663
- —EU ‐ INF‐ACT Cascade Open Call 2023 (COC‐1‐2023‐CNR)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsOrganic Chemistry Synthesis Methods · Radical Photochemical Reactions · Cyclopropane Reaction Mechanisms
Introduction
1
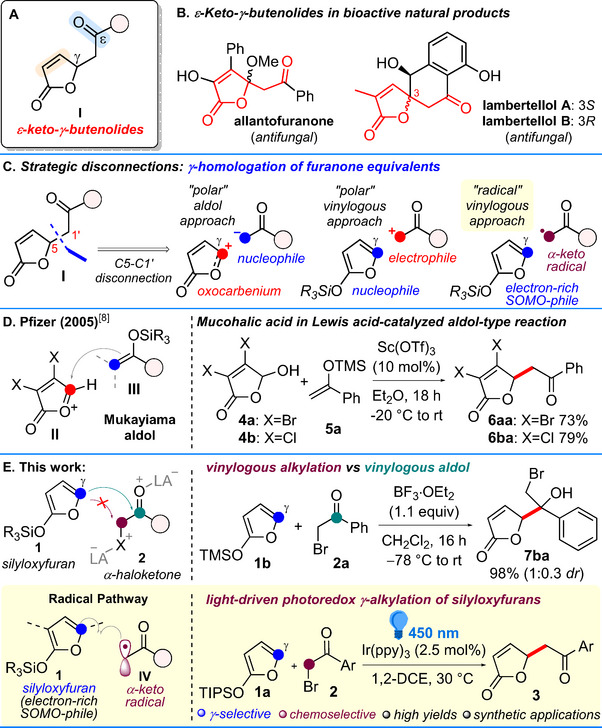
γ‐Butenolides—i.e., substituted furan‐2(5H)‐ones—are five‐membered unsaturated lactones which constitute the structural core of a wide array of natural and unnatural products displaying an impressive range of biological activities in the fields of pharmaceuticals, agrichemicals, and organic materials [1, 2]. A peculiar, yet underestimated butenolide subset comprises ε‐keto‐γ‐butenolides of type I (Scheme 1A) which feature an ε‐ketone side chain linked to the stereogenic C5(γ)‐position of the α,β‐unsaturated lactone moiety. Such chiral, tridentate electrophiles, apart from being pivotal “per se” in several bioactive natural products, as antifungal allantofuranone [3] and lambertellols A and B [4, 5] (Scheme 1B), might also serve as prominent platforms for late‐stage modifications to build novel, complex, heterocyclic derivatives (vide infra).
(A) General structure of the targeted ε‐keto‐γ‐butenolides of type I. (B) Examples of natural products embedding the ε‐keto‐γ‐butenolide framework. (C) Strategic C5‐C1′ disconnections toward I. (D) Aldol‐type approach by J. Zhang et al. at Pfizer (see Ref. [8]). (E) This work: preliminary investigation on the feasibility of the polar, vinylogous alkylation strategy (see Section 3 of the Supporting Information for details), and outline of the successful radical approach.
Quite unexpectedly though, efficient synthetic methodologies enabling the construction of these architectures are quite underdeveloped, still limited almost exclusively to phthalide derivatives [6, 7]. From a synthetic point of view, one of the most straightforward access to the ε‐keto‐γ‐butenolide framework entails the strategic disconnection at the C5‐C1′ bond of I which envisages the regioselective γ‐homologation of a furan‐2(5H)‐one equivalent with a coupling (aryl)acyl partner (Scheme 1C). In this context, a first elegant approach was reported by Pfizer in 2005 [8], involving the Lewis acid‐catalyzed, Mukaiyama aldol‐type reaction between mucohalic acids 4a and 4b and various silyl ketene acetals or silyl enol ethers (Scheme 1D). In that instance, treatment of 4a and 4b with Sc(OTf)3 (10 mol%) and the silyl enol ether from acetophenone 5a, provided the halogenated 5‐phenacylmethyl γ‐butenolides 6aa and 6ba in good, isolated yields via formation of the strongly electrophilic oxocarbenium ion II at the C‐γ of the butenolide. A reversed option would entail the vinylogous γ‐alkylation of a silyloxyfuran of type 1 with α‐haloketones of type 2 as suitable alkylating agents (Scheme 1E). Interestingly, despite the apparent easiness of this strategy, the γ‐alkylation of vinylogous silyloxyfurans 1 via a S* N 1 or S N *2 mechanism on haloketones of type 2 is still unprecedented. Indeed, the chemoselective substitution of a nucleophile to the C(sp ^3^)−X of 2 appears challenging, as its bidentate electrophilic nature makes the aldol addition at the carbonyl a viable, competitive option [9]. To shed light onto the feasibility of this transformation, in this work we preliminarily tested a series of Lewis acids to promote the vinylogous alkylation of trimethylsilyloxyfuran 1b with phenacyl bromide 2a (Scheme 1E; see also Table S1 in the Supporting Information) [10]. Under these conditions, we observed the formation of the sole aldol adduct 7ba, instead of the expected alkylated product, which was detected but only in traces in the reaction crude mixtures. On this ground, having established the vinylogous aldol reactivity bias of 1 with 2 in the polar domain, we turned to evaluate the viability of this alkylation according to a radical approach. Radicals have emerged as a powerful tool in advanced organic synthesis, offering unique reaction mechanisms and a wide range of applications [11, 12]. In this context, an efficient and sustainable method to promote radical reactions is visible‐light photoredox catalysis, which has polarized widespread research interest owing to its attractive features such as mild conditions, excellent functional group tolerance, and high reactivity [13, 14, 15]. Considering the strategic disconnection described in Scheme 1C, α‐haloketones 2, like phenacyl bromide 2a, are well known radical precursors which enable the formation of the corresponding α‐keto radical IV intermediates by single electron transfer (SET) reduction of the C─Br bond. As an electrophilic radical, intermediate IV could readily couple with electron‐rich olefins (SOMO‐philes) like dienamines [16], silyl enol ethers [17, 18, 19], or their vinylogous π‐extended congeners [20]. We envisaged that, in principle, radical IV could likely be trapped by electron‐rich silyloxyfurans 1, hence delivering the otherwise hardly accessible γ‐butenolides of type 3 (Scheme 1E, bottom). Continuing our efforts toward the discovery of novel synthetic methodologies, that would merge radical chemistry and the principle of vinylogy to expand the structural domain currently accessible through the common (poly)enolate chemistry [21], we here introduce a novel, chemoselective and γ‐regioselective methodology to afford ε‐keto‐γ‐butenolides 3, in one single step and high efficiency. Furthermore, late‐stage functionalization of scaffolds 3 has been accomplished, demonstrating their usefulness as versatile platforms to readily access complex, heterobicyclic derivatives.
Results and Discussion
2
At the outset of our investigation, we selected triisopropylsilyloxyfuran (TIPSOF) 1a and phenacyl bromide 2a as model substrates to evaluate the proof‐of‐concept and to optimize reaction conditions (Table 1). Initially, we began our survey by testing conditions common to similar transformations [20], i.e., using fac‐Ir(ppy)3 (Ir‐1) as the photocatalyst (2.5 mol%), K_2_CO_3_ (2 mol equivalents) in degassed CHCl_3_ under blue LED irradiation (450 nm), at 30°C for 5 h. Under these conditions, 1a reacted with excess 2a (1.5 equiv) to produce the desired product 3aa (35% yield after column chromatography), along with the unexpected γ,γ‐bis‐alkylated adduct 8aa in a 1:0.17 mixture (Table 1, entry 1). Evaluation of several other metal‐ and organic photocatalysts did not improve the formation of 3aa, with Ir‐2 and Ru‐1 being the only alternatives, although giving quite lower yields compared to Ir‐1 (entries 2 and 3 and Table S2 in the Supporting Information). We then surveyed other solvents, identifying 1,2‐dichloroethane (1,2‐DCE) as a better alternative (entry 4), which provided 3aa in a 42% isolated yield, accompanied by a lower amount of 8aa (1:0.1 3aa:8aa).
TABLE 1: Optimization of reaction conditions (selected entries). a
Envisaging that the formation of 8aa could be fostered by the carbonate [22], we tested the reaction without the base in a more dilute solution (Table 1, entry 7); pleasingly, after 5 h at 30°C, 3aa was isolated in an improved 50% yield, accompanied by a corresponding decrease of 8aa (1:0.06 3aa:8aa). The real breakthrough occurred by switching the stoichiometric ratio between 1a and 2a to 1.5:1 (entries 8 and 9), ultimately providing 3aa with an optimal 93% isolated yield after 3 h, with only traces of 8aa (entry 9). Finally, the use of the less hindered trimethylsilyloxyfuran (TMSOF) 1b proved to be almost equally productive as 1a, providing 3aa in a 88% isolated yield while reducing the generation of 8aa (1:0.01 3aa:8aa, entry 10). Importantly, in all cases, full γ‐regioselectivity was observed, with no detection of α‐alkyl products. Likewise, no products deriving from attack to the ketone moiety were observed, testifying complete chemoselectivity. Finally, as evidence of its photocatalytic nature, the reaction was completely inert in the absence of either the photocatalyst (entry 11) or light (entry 12). Having established the optimal reaction conditions for accessing γ‐alkylated butenolide 3aa from 1a or 1b (Table 1, entries 9 and 10), we proceeded to evaluate the scope and limitations of the process. As depicted in Table 2, a series of differently substituted aromatic and aliphatic α‐bromoketones 2a‐r underwent the desired transformation furnishing the corresponding ε‐keto‐γ‐butenolides 3.
TABLE 2: Substrate scope of the photoinduced γ‐alkylation of silyloxyfurans 1a‐d. a , b
Concerning the reactivity of aromatic ketones (acetophenones) with TIPSOF 1a, irrespective of the electron‐withdrawing or electron‐donating ability of the substituents on the aryl ring, the reaction proved viable under the optimized reaction conditions, as demonstrated, for example, by the electron‐rich 4‐methoxy congener 2f and the electron‐poor 4‐cyano‐acetofenone 2g which provided the best results, delivering the corresponding adducts 3af and 3ag in highly rewarding yields. Several compounds, including electron‐poor ortho‐fluoro‐acetophenone 2k and para‐ and meta‐trifluoromethyl‐acetophenones 2h and 2i reacted better with the less hindered TMSOF 1b, providing compound 3ak in a good 82% isolated yield, and butenolides 3ah and 3ai in slightly lower yields around 70%. An exception was observed with electron‐deficient ketones 2q and 2r, bearing nitro groups at the para‐ and meta‐positions, respectively, which failed to provide the expected adducts in satisfactory yields (<20% isolated yield for both). In contrast, 1b proved to be a competent substrate for sterically demanding secondary and tertiary bromides 2m and 2n. Under the optimized conditions, the prostereogenic acetophenone 2m reacted with 1b to furnish the corresponding γ‐adduct 3am in a good 73% isolated yield as a 1:1 syn/anti mixture. An unusual outcome was observed with tertiary 2‐bromo‐2‐methyl‐acetophenone 2n, which afforded a 1.3:1 γ/α regioisomeric mixture of products 3an in a combined 70% yield (40% isolated yield for the sole γ‐adduct) [23, 24]. Finally, γ‐ and α‐methyl‐substituted silyloxyfurans 1c and 1d were also tested. Under the optimized reaction conditions, both scaffolds reacted with 2a with similar efficiency, affording the corresponding γ‐adducts 3ca and 3 da in 55% and 65% yields, respectively. As a proof of the generality of the disclosed procedure, the reaction also proceeded smoothly with aliphatic bromides such as 1‐bromo‐2‐butanone 2o and 1‐bromopinacolone 2p, which reacted with 1b to deliver the γs‐adducts 3ao and 3ap in 50% and 75% isolated yields, respectively.
The reaction between 1b and 2a was successfully carried out also on a gram‐scale using 5.0 mmol of 2a. Gladly, under slightly modified reaction conditions (i.e., 465 nm blue LED strip source, and 0.07 M instead of 0.06 M reaction concentration; see Section 4.4 of the Supporting Information for details), pure 3aa was obtained (0.86 g, 85% isolated yield) without any loss of regioselectivity (Scheme 2a). Of note, subjecting the reaction mixture to vacuum‐driven evaporation for 12 h, provided a crude with the sole 3aa in a 90% NMR purity (0.91 g, 90% yield). As a further extension of the scope, the disclosed methodology could also be employed to regioselectively functionalize more extended π‐systems, alias hypervinylogous systems [25], such as electron‐rich, methyl‐ and tert‐butyl‐substituted γ‐vinyl silyloxyfurans 1e and 1f (Scheme 2b). Under the optimized reaction conditions, both trienes were able to trap the α‐ketoradical derived from 2a selectively at the most distal ε‐site, providing η‐keto‐γ‐vinylidene butenolides 9ea and 9fa as 1:1 mixture of the corresponding Z/E isomers in 60% and 45% combined yields, respectively.
(a) Gram‐scale experiment conducted with a 465 nm blue LED strip source (14.4 W) (b) Photoinduced ε‐alkylation of extended silyloxyfurans 1e and 1f. Reaction conditions: 1 (1.5 equiv), 2a (0.1 mmol, 1.0 equiv), Ir‐1 (2.5 mol%) in degassed 1,2‐DCE [0.06 M] at 30°C, irradiated with blue LEDs (450 nm) for 3 h. Yields refer to isolated yields of 9 after silica gel flash chromatography. The Z/E ratio was determined by 1H NMR analysis of the crude. See the Supporting Information for details.
Having established an efficient procedure for accessing chiral ε‐keto‐γ‐butenolide chemotypes of type 3, we then moved on to demonstrate their versatility as strategic platforms to forge fused heterobicyclic scaffolds such as tetrahydrofuro[3,2‐b]furan‐2(3H)‐ones of type 10 [26] (Scheme 3b‐c), and the tetrahydrofuro[3,2‐c]pyridazine‐6(1H)‐one 13 (Scheme 3d) [27, 28, 29]. To this end, we envisaged a two‐step strategy centered upon the interconversion of the ε‐ketone moiety of I (e.g. by reduction to secondary alcohol or hydrazone formation), followed by ring closure at the β‐position of the electron‐deficient unsaturated lactone (Scheme 3a).
(a) General strategy for late‐stage modification of butenolides I. (b) One‐pot access to bicyclic furanones (±)‐10 and (±)‐epi‐10. (c) Two‐step access to (±)‐10 via dynamic kinetic resolution (DKR) of diastereomers syn‐11 and anti‐11. (d) One‐pot access to bicyclic pyridazine‐6(1H)‐one (±)‐13. In all cases yields refer to isolated yields; the ratio 10/epi‐10 was determined by 1H NMR of the crude. See the Supporting Information for details.
Indeed, treating 3aa with slight excess NaBH_4_ in MeOH (at −15°C to 25°C for 24 h) failed to deliver the expected open‐chain alcohol, ending up in a 2:1 mixture of the cis‐fused dioxabicycles (±)‐10 and (±)‐epi‐10, in a good 70% combined, isolated yield (Scheme 3b). The easiness of this one‐pot, ketone reduction/oxa‐Michael cyclization cascade prompted us to investigate a more diastereoselective procedure to access either (±)‐10 or (±)‐epi‐10. After a brief survey (see Table S8 in the Supporting Information for details), we found that treatment of 3aa with NaBH_4_ (1.1 equiv) in a more concentrated MeOH solution [16 mM], for 1 h, produced alcohol 11 as a racemic 1:1 syn/anti mixture in a combined 72% yield. Quite unexpectedly though, treating this equimolar mixture of diastereomers with a sub‐stoichiometric amount of DBU (0.5 equiv) in THF under dilute conditions [6 mM] enabled the formation of 5‐phenyltetrahydrofuro[3,2‐b]furan‐2(3H)‐one (±)‐10 in a good 65% isolated yield (47% overall) as an almost single isomer (1:0.08 10/epi‐10, Scheme 3c). This result is far from being trivial, since it implies the diastereoselective cycloetherification of the 1:1 syn/anti mixture of alcohol 11 via a peculiar dynamic kinetic resolution (DKR) [30, 31, 32]. Such DKR is likely enabled by the DBU‐catalyzed, vinylogous C5 site epimerization [33] of syn‐11 into anti‐11, which cyclizes more rapidly than its isomer, thus selectively leading to (±)‐10 (Scheme 3c). An aza‐version strategy was also implemented, involving the formation of a hydrazone and its subsequent intramolecular aza‐Michael ring closure. Indeed, treating 3aa with phenylhydrazine (12) in the presence of a catalytic amount of glacial acetic acid (Scheme 3d) [17] directly produced a single, heterobicyclic compound, namely 1,3‐diphenyl‐4,4a,7,7a‐tetrahydrofuro[3,2‐c]pyridazin‐6(1H)‐one (±)‐13 in one‐pot and in a good 65% isolated yield (Scheme 3d).
Based on previous studies highlighting the ability of electron‐rich polyenes to trap electrophilic radicals at distal positions [20, 34, 35, 36], and a series of control proofs such as TEMPO trapping, and On‐Off experiments (see Sections 6.1 and 6.2 in the Supporting Information for details), a plausible catalytic cycle for the photoredox γ‐alkylation of 2‐silyloxyfurans is proposed, as exemplified by the addition of 2a to 1a (Scheme 4). The reaction is initiated with the well‐known single‐electron reduction of phenacyl bromide 2a [E ox = −0.49 V vs. Ag/AgCl] [34, 37] by the excited state photocatalyst *Ir^III^(ppy)3 [E ox = −1.73 V vs. SCE] [15] which affords the corresponding α‐keto radical 2a ^•^.
Proposed catalytic cycle.
Such an electrophilic radical is then selectively trapped at the γ‐position of the electron‐rich diene 1a, forming the radical intermediate 3aa ^•^, which is subsequently oxidized to the corresponding carbocation **3aa^+^ ** via reduction of the newly formed Ir^IV^(ppy)3 [E red = +0.77 V vs. SCE] [15] hence closing the photocatalytic cycle. Finally, desilylation of **3aa^+^ ** by the bromide anion results in the formation of the observed adduct 3aa [22].
The pronounced γ‐regioselectivity observed in nearly all reactions examined in this study highlights the strong bias of electron‐rich polyenes such as 1a and 1b to preferentially “trap” electrophilic radicals at remote positions [20, 35, 36]. From a theoretical perspective, this site selectivity is likely governed by two factors: (i) a favorable overlap between the HOMO of the diene system 1 and the SOMO of the electrophilic radical **2^•^ **, and (ii) the formation of a stabilized oxyallyl radical intermediate **3aa^•^ ** (Scheme 4), which is uniquely accessible through the γ‐addition pathway. As a support of this rationale, we took into consideration the DFT calculations performed in a very recent work, aiming at elucidation of the electronic distribution of 1a and 1b, based on HOMO composition and global/local electrophilicity and nucleophilicity indices [21]. In that study, the neutral, closed‐shell 1a exhibited a pronounced nucleophilic character, consistent with its ability to trap electrophilic radicals and carbocations. Both HOMO orbital analysis and condensed Fukui indices (f^−^) confirmed a strong preference for γ‐addition, a trend also observed for 1b.
Conclusion
3
In conclusion, we have successfully developed a light‐mediated, γ‐regioselective alkylation of 2‐silyloxyfurans 1 with α‐bromoketones 2, by exploiting a photoredox catalytic cycle promoted by blue light. This approach provides practical and efficient access to chiral, ε‐ketobutenolides of type 3, in one single step, with high efficiency and complete γ‐regiocontrol. The usefulness of these scaffolds as versatile platforms to access complex, heterobicyclic derivatives was also demonstrated. Building on our investigations on the reactivity of silyloxyfurans in the field of vinylogous, photo‐promoted radical reactions, we anticipate that this methodology will inspire future discoveries for the direct and remote functionalization of the butenolide core in natural products and medicinal chemistry. Indeed, as this work demonstrates, the combination of visible light‐mediated radical chemistry and the peculiar reactivity of vinylogous systems could be used to promote elusive transformations not accessible via conventional “polar” pathways, enabling access to novel chiral chemotypes. Further investigations aimed at implementing enantioselective pathways for similar transformations, and the evaluation of the antiviral activity of heterobicycles (±)‐10, (±)‐epi‐10 and (±)‐13 are currently ongoing in our laboratory.
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
Supporting File 1: The authors have cited additional references within the Supporting Information.
Supporting File 2: chem70577‐sup‐0002‐DataFile.pdf
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1A. Husain , S. A. Khan , F. Iram , M. A. Iqbal , and M. Asif , “Insights Into the Chemistry and Therapeutic Potential of Furanones: A Versatile Pharmacophore,” European Journal of Medicinal Chemistry 171 (2019): 66–92, 10.1016/j.ejmech.2019.03.021.30909021 · doi ↗ · pubmed ↗
- 2B. Mao , M. Fañanás‐Mastral , and B. L. Feringa , “Catalytic Asymmetric Synthesis of Butenolides and Butyrolactones,” Chemical Reviews 117 (2017): 10502–10566, 10.1021/acs.chemrev.7b 00151.28640622 PMC 5553117 · doi ↗ · pubmed ↗
- 3A. Schüffler , D. Kautz , J. C. Liermann , T. Opatz , and T. Anke , “Allantofuranone, a New Antifungal Antibiotic From Allantophomopsis Lycopodina IBWF 58B‐05A,” The Journal of Antibiotics 62 (2009): 119–121, 10.1038/ja.2008.21.19198630 · doi ↗ · pubmed ↗
- 4M. Nomiya , T. Murakami , N. Takada , T. Okuno , Y. Harada , and M. Hashimoto , “Syntheses of Lambertellols and Their Stable Analogues; Investigation of the Real Active Species in the Mycoparasitism by Lambertella Species,” The Journal of Organic Chemistry 73 (2008): 5039–5047, 10.1021/jo 8005478.18522417 · doi ↗ · pubmed ↗
- 5T. Murakami , Y. Morikawa , M. Hashimoto , T. Okuno , and Y. Harada , “Lambertellols A and B, Novel 3,4‐Dihydronaphthalen‐1(2 H )‐ones with Spiro‐Butenolide Produced by Lambertella sp.1346,” Organic Letters 6 (2004): 157–160, 10.1021/ol 035889 d.14723517 · doi ↗ · pubmed ↗
- 6W.‐H. Rao , Q. Li , Y.‐G. Li , et al., “Visible‐Light Photoredox‐Catalyzed Acyl Lactonization of Alkenes with Acyl Chlorides,” European Journal of Organic Chemistry 26 (2023): e 202300191, 10.1002/ejoc.202300191. · doi ↗
- 7X. Liang , M. Xiong , H. Zhu , K. Shi , Y. Zhou , and Y. Pan , “Copper/Palladium Bimetallic System for the Synthesis of Isobenzofuranones Through [4 + 1] Annulation Between Propiophenones and Benzoic Acids,” Organic Letters 22 (2020): 9568–9573, 10.1021/acs.orglett.0c 03627.33284633 · doi ↗ · pubmed ↗
- 8P. Angell , J. Zhang , D. Belmont , T. Curran , and J. G. Davidson , “Mucohalic Acid in Lewis Acid Catalyzed Mukaiyama Aldol Reaction: A Concise Method for Highly Functionalized γ‐Substituted γ‐Butenolides,” Tetrahedron Letters 46 (2005): 2029–2032, 10.1016/j.tetlet.2005.01.147. · doi ↗