Disease-modifying therapies for Rett syndrome: a review for neurologists
Debopam Samanta

TL;DR
This paper reviews disease-modifying therapies for Rett syndrome, focusing on gene therapy approaches and their implications for neurologists.
Contribution
The paper provides a detailed review of the most advanced gene replacement therapies for Rett syndrome and highlights translational challenges.
Findings
TSHA-102 and NGN-401 show early efficacy in clinical trials for Rett syndrome.
Gene therapy for Rett syndrome faces challenges like expression toxicity and X-chromosome inactivation.
Next-generation strategies include CRISPR-Cas9 and RNA editing for potential mutation correction.
Abstract
Rett syndrome (RTT) is a severe X-linked neurodevelopmental disorder affecting approximately 1 in 10,000–15,000 females, most often caused by loss-of-function mutations in MECP2. Until the recent approval of trofinetide, management relied exclusively on symptomatic treatment and multidisciplinary supportive care. The therapeutic landscape is now undergoing a rapid shift, driven by multiple gene therapy approaches designed to restore functional MeCP2 expression and achieve true disease modification. As these therapies progress toward potential regulatory approval, neurologists will play central roles in identifying eligible patients, counseling families, supporting clinical trial enrollment, delivering treatments, monitoring long-term outcomes, and advocating for equitable access. This review provides neurologists with the essential framework needed to understand and navigate this…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
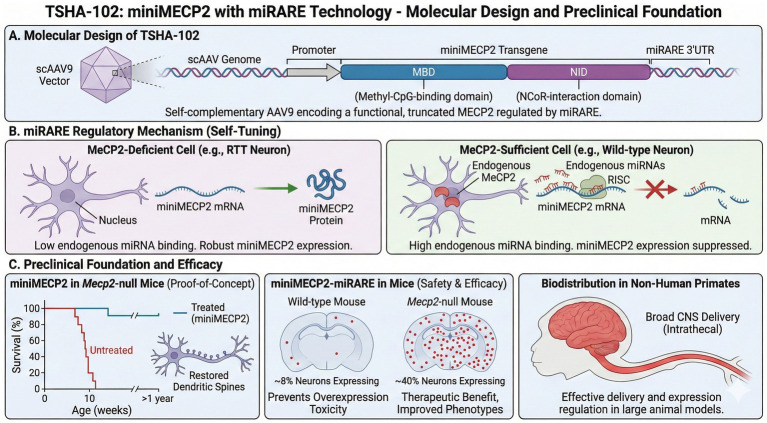 Figure 1
Figure 1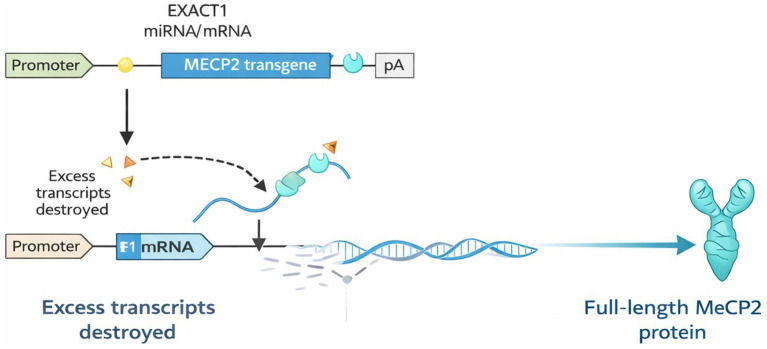 Figure 2
Figure 2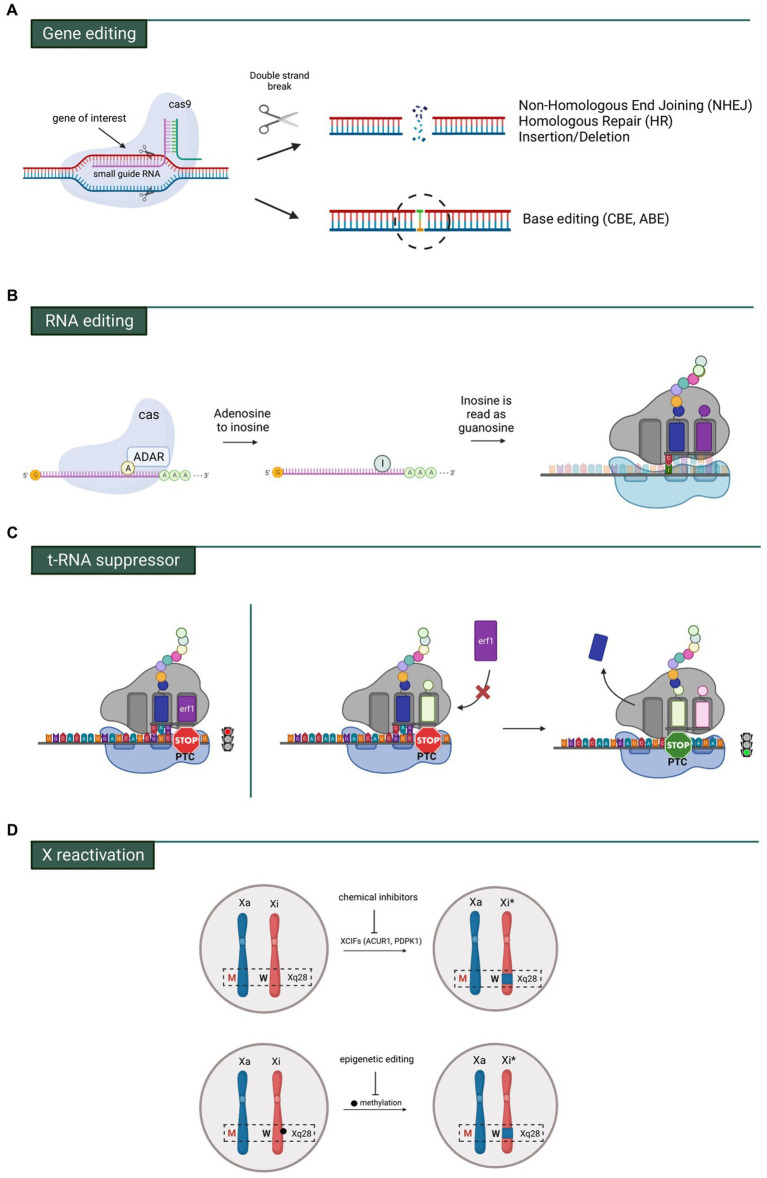 Figure 3
Figure 3| Stage | Age range | Key features | Neurologic/Motor features | Behavioral features | Other features |
|---|---|---|---|---|---|
| I. Early Onset/Stagnation | 6–18 months | Subtle developmental slowing | Hypotonia; decreased eye contact | Reduced exploration | Deceleration of head growth |
| II. Rapid Destructive | 1–4 years | Rapid regression of skills | Loss of purposeful hand use; stereotypic hand movements; loss of language; gait apraxia/ataxia | Irritability; inconsolable crying; social withdrawal | Irregular breathing patterns; onset of seizures |
| III. Plateau/Pseudostationary | 2–10 + years | Stabilization of regression | Persistent apraxia, ataxia; worsening scoliosis | Improved eye gaze and nonverbal communication | Growth retardation; peak seizure burden |
| IV. Late Motor Deterioration | Late childhood to adulthood | Progressive motor decline | Loss of ambulation; increasing rigidity, spasticity, dystonia | Behavioral improvement; fewer seizures | Increased risk of fractures and immobility complications |
| Category | Criteria |
|---|---|
| Main Criteria (all required for typical RTT) |
Loss of purposeful hand skills Loss of spoken language Gait abnormalities (ataxia, apraxia, or loss of ambulation) Stereotypic hand movements |
| Supportive Criteria (≥5 required for atypical RTT) |
Breathing irregularities while awake Bruxism Sleep disturbances Abnormal muscle tone Peripheral vasomotor disturbances Scoliosis/kyphosis Growth retardation Small, cold hands and feet Inappropriate laughing or screaming spells Diminished pain response Intense eye communication |
| Required for molecular confirmation | Documented pathogenic MECP2 variant |
| System/concern | Recommended evaluation | Comments | Management |
|---|---|---|---|
| Constitutional | Height, weight, head circumference | Baseline growth and nutritional status | Nutrition optimization; monitor for growth failure |
| Neurologic | Neurology exam, brain MRI; EEG/video EEG if seizures suspected | High seizure prevalence; evaluate tone/movement | ASM management; seizure safety education |
| Development | Developmental, motor, cognitive, speech-language evaluation | Determines therapy needs | Early intervention (0–3 yr), special education (3 + yr), PT/OT/SLP |
| Psychiatric/Behavioral | Neuropsychiatric screening (>12 months): sleep, ADHD, anxiety, ASD traits | Behavioral features common | Melatonin for sleep; SSRIs/risperidone for agitation/anxiety |
| Musculoskeletal | Ortho/PM&R; PT/OT assessment; scoliosis screening | High risk of scoliosis and mobility decline | PT/OT, bracing, positioning devices; surgical referral if needed |
| Gastrointestinal/Feeding | GI/nutrition evaluation; aspiration risk assessment | Constipation, reflux, feeding difficulty common | Constipation regimen; GERD therapy; feeding therapy; consider G-tube |
| Respiratory | Overnight sleep study | Evaluates apnea, hypoventilation | Respiratory support as needed; sleep interventions |
| Sleep disorders | Portable polygraphic screening | Detects apnea/hypopnea | Melatonin ± hydroxyzine/diphenhydramine |
| Cardiovascular | Cardiology evaluation, ECG | Risk of prolonged QTc | Avoid QT-prolonging drugs; cardiology follow-up |
| Osteopenia | Bone densitometry | High fracture risk | Optimize Ca/Vit D; encourage weight-bearing; follow bone health guidelines |
| Vision | Ophthalmology evaluation | Strabismus, visual impairment common | Vision therapy, school-based services |
| Hearing | Audiology evaluation | Rule out hearing loss | Hearing aids; educational supports |
| Genitourinary | Clinical evaluation | Functional incontinence common | Urology/pelvic floor referral as needed |
| Integument/Autonomic | Skin/vascular exam | Cold, poorly perfused hands/feet | Warmth strategies; monitor autonomic symptoms |
| Genetics | Geneticist/genetic counselor consult | Family counseling | Ongoing counseling; recurrence-risk discussion |
| Family resources | Social work; community support needs | High caregiver burden | Respite, home nursing, palliative care when appropriate |
| Study (Name/publication) | Dose/Regimen | Duration | Efficacy/Key findings | Adverse effects/Safety | |
|---|---|---|---|---|---|
| LAVENDER (Phase 3 RCT), Neul et al. ( | 187 (93 trofinetide/94 placebo); age 5–20 yrs. | Weight-based trofinetide, twice daily (oral) | 12 weeks |
Co-primary endpoints: RSBQ change −4.9 vs. − 1.7 ( Key secondary: communication/social (CSBS-DP-IT Social) improved (LS mean difference 1.0, | Diarrhea 80.6% vs. 19.1% placebo (mostly mild–moderate), vomiting 26.9% vs. 9.6% placebo. 17.2% discontinuation due to TEAEs in trofinetide arm vs. 2.1% placebo. Serious AEs 3.2% in both groups. |
| DAFFODIL (Phase 2/3 open-label) — Percy et al. ( | 15 girls; age 2–4 yrs. | Weight-based trofinetide, bid (oral) | Period A 12 weeks + longer-term follow-up (~21 months) | Exploratory efficacy: improvements in CGI-I, caregiver-reported global impression (CaGI-I), and quality of life (ICND-QoL). Caregiver exit interviews ( | Treatment-emergent AEs: diarrhea 80%, vomiting 53.3%, mostly mild/moderate. PK data: weight-based dosing achieved target exposure. |
| LILAC (open-label extension of LAVENDER) — 2024 report | 154 females; age 5–21 yrs. | Trofinetide, bid, open-label (oral) | 40 weeks | RSBQ mean change from LAVENDER baseline to week 40: −7.3 (SE 1.62) in those originally on trofinetide, −7.0 (SE 1.61) in those switched from placebo. CGI-I at week 40 (from LILAC baseline): 3.1 (SE 0.11) for prior trofinetide, 3.2 (SE 0.14) for prior placebo. | AEs: diarrhea 74.7%, vomiting 28.6%, COVID-19 11%. Diarrhea caused withdrawal in 21.4%. No new safety signals beyond those in LAVENDER. |
| LILAC-2 (32-month open-label extension) — Percy et al. ( | 77 females; age 5–22 yrs. (mean ~12.0 ± 4.4 yr) | Trofinetide, bid, open-label (oral) | Up to 32 months (median exposure ~811 days) | Sustained improvement: RSBQ change from LAVENDER baseline to week 104 of LILAC-2: −11.8 (SE 2.45). CGI-I at week 12 of LILAC-2 (from LILAC baseline): 3.1 (SE 0.10). Most caregivers (96%) reported being “satisfied” or “very satisfied” with treatment benefits. | AEs: diarrhea 53.2%, vomiting 19.5%, COVID-19 27.3%. No new safety signals. |
| LOTUS (real-world, observational, up to 12 months) — interim data 2025 | 227 patients (US, pediatric & adult), on trofinetide under routine care; age ≥2 yrs. (per approval criteria) | Weight-based, bid, oral or via gastrostomy tube per real-world prescription | Up to 12 months (interim) | Median dose at week 1: 36% of target, increased to >80% by week 10; 55.2% reached full recommended daily dose Caregiver-reported behavioral improvements (BIQ) months 1–12: Non-verbal communication 48–71%, Alertness 44–70%, Social interaction & connectedness 33–58% Median QI-Disability total score change from baseline: 4.7 → 4.6 (months 1–12) |
Diarrhea: weeks 1–12 (23–50%), months 4–12 (26–38%), mostly contained in diapers Safety consistent with prior trofinetide trials |
| Product name | Proposed mechanism of action | Current status in research |
|---|---|---|
| 2–73 (blarcamesine) | Sigma-1 receptor activator | Phase 2/3 study in girls (5–17 years) completed in Australia, Canada, UK - did not meet statistical significance on primary endpoints at 12 weeks. Phase 3 adult study completed in Australia/UK with topline results expected soon. Has FDA Fast Track, Rare Pediatric Disease, and Orphan Drug designations. |
| Fenfluramine | Serotonin 5-HT2 receptor agonist | Phase 3 clinical program planned by UCB Pharma, expected to begin first half of 2026. Already FDA-approved for Dravet and Lennox–Gastaut syndromes. |
| Leriglitazone | Selective PPAR- | Phase 2a TREE Study initiated March 2025 in Spain. Placebo-controlled, 36-week study in 24 pediatric patients (up to 17 years) with confirmed MECP2 mutations. |
| NTI164 | Broad-spectrum medical cannabinoid | Phase I/II trial in Australia. After 12 weeks (May 2024), 93% of participants showed improvement on CGI-I scale with 205% mean improvement on RSBQ from weeks 4–12. All participants enrolled in 52-week extension. Has FDA Orphan Drug Designation (Nov 2024), EMA Orphan Drug Designation (March 2025), and FDA Rare Pediatric Disease Designation (Oct 2025). |
| Vorinostat (RVL-001) | Small molecule oral therapeutic (mechanism specific to Rett not detailed) | Clinical trial material manufacturing initiated in 2024 for trials in US and Colombia. Proof-of-concept “n-of-1” placebo-controlled trial planned for early summer 2025 in Colombia with 15 patients. Has FDA Orphan Drug Designation (May 2024). |
| DPM-1003 | Protein tyrosine phosphatase (PTP) family enzyme inhibitor | FDA clearance received March 2024 to initiate Phase 1 clinical trial. Has Orphan Drug designation and conditional Rare Pediatric Disease designation. |
| MB-204 | Adenosine A2A receptor antagonist; modified version of istradefylline (FDA-approved for Parkinson’s) | Preclinical stage. Mouse study data released March–April 2025 showed greater improvement in social behaviors vs. trofinetide. Plans to file for FDA Orphan Drug Designation announced March 2025. |
| NQ-13 | Peptide-based IGFBP2 mimetic; involved in neurogenesis, synaptic function, and cognition | Preclinical development. In rat model, reversed movement, breathing, and cognitive dysfunction, and restored brain MeCP2 levels to normal values. |
| AMO-04 | Glutamate modulator | Preclinical development. Granted FDA Orphan Drug Designation in June 2018. Showed promise in mouse model through IRSF’s Scout Program. |
| KIT-13/KIT-14 | Synthetic plasmalogen derivative; anti-inflammatory effects via p65 nuclear accumulation inhibition | Preclinical development. KIT-13 granted FDA Rare Pediatric Disease and Orphan Drug Designations in March 2023. KIT-14 received EMA Orphan Drug Designation in June 2025. |
| NLX-101 | Serotonin 5-HT1A receptor activator | Preclinical development. Published study in March 2025 showed improvements in respiratory impairment and cognitive deficits in Rett mouse model. Reduced apneas and normalized irregular breathing patterns. |
| OV4041 | Direct KCC2 activator; restores excitatory/inhibitory balance and supports GABA inhibition | Preclinical stage. Plans announced June 2025 to begin preclinical studies in late 2025 for IND filing, with IND submission expected late 2026. Has anxiolytic, antipsychotic, and anticonvulsant activity. |
| Axonis Therapeutics (TBD) | KCC2-targeting medicines to maintain inhibitory signaling in the brain | Preclinical research. Collaborative grant with Tang Lab at Boston Children’s Hospital. Research showed increasing KCC2 expression can rescue cellular and behavioral abnormalities in Rett. |
| Pridopidine | Sigma-1 receptor (S1R) agonist with neuroprotective properties | Preclinical evaluation ongoing by Prilenia Therapeutics. Currently in late-stage development for Huntington’s disease and ALS. |
| Gliachem compounds | TRPM2 channel blockers; boost mTOR activity to restore brain development and function | Preclinical development. New molecules designed to block TRPM2, which negatively regulates mTOR and is over-expressed in Rett brain. |
| Palena Therapeutics EERPs | Immunomodulatory peptides (embedded epitope random peptides) that increase BDNF expression | Preclinical concept stage. Similar mechanism to Copaxone in MS. Small open-label study of Copaxone (n = 10) showed improvements in gait, visual attention, memory, and breath holding index. |
| Fine motor/Hand function | Gross motor | Communication/Social |
|---|---|---|
| Reached for toy | Sat with support when placed | Responded to familiar names or words |
| Taken a drink from a cup held without assistance | Sat without support when placed | Followed a command with a gesture |
| Used raking grasp to retrieve an object | Came to sitting | Followed a command without a gesture |
| Used a pincer grasp (either refined or modified) | Pulled to standing | Pointed for something they want |
| Finger fed | Stood while holding on | Waved bye-bye |
| Transferred an object from one hand to the other | Stood independently | Babbled |
| Used a spoon/fork to eat without assistance | Cruised around furniture or while holding someone’s hand | Used words with meaning |
| Walked independently | Spoken in phrases (two words or more with meaning) | |
| Climbed up stairs with help | ||
| Climbed up stairs without help | ||
| Climbed down stairs with help | ||
| Climbed down stairs without help | ||
| Ran 10 feet without falling |
| Feature | TSHA-102 | NGN-401 |
|---|---|---|
| MECP2 construct | Truncated miniMECP2 (MBD + NID domains only) | Full-length human MECP2 |
| Regulatory system | miRARE (endogenous miRNA-responsive) | EXACT (synthetic miRNA feedforward loop) |
| AAV type | Self-complementary AAV9 | Standard single-stranded AAV9 |
| Administration route | Intrathecal lumbar puncture | Intracerebroventricular (ICV) |
| Procedural complexity | Moderate (lumbar puncture) | Higher (neurosurgical ICV access) |
| CNS biodistribution | Moderate (based on IT delivery) | Superior (10–100 × higher in preclinical NHP studies) |
| Clinical stage | Phase 1/2 Part A complete; Part B pivotal enrolling | Phase 1/2 transitioning to pivotal; Arm 4 enrolling |
| Safety profile to date | Favorable; no SAEs or DLTs | Favorable at low doses; high-dose hyperinflammatory fatality |
| Efficacy signals | All participants gained ≥1 milestone; sustained improvements | Low-dose: 23 skills across 4 participants; sustained gains |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenetics and Neurodevelopmental Disorders · CRISPR and Genetic Engineering · Autism Spectrum Disorder Research
Introduction
1
Rare diseases collectively affect a surprisingly large segment of the population, with an estimated 3.5–5.9% of individuals worldwide living with one of more than 6,000 recognized conditions—most of which are genetic and begin in childhood (1). Rare neurodevelopmental disorders represent a substantial portion of this burden and, although individually uncommon, they represent a major component of pediatric and adult neurology practice. Neurologists therefore serve as the primary long-term physicians for many of these patients, guiding diagnostic workup, symptom management, and multidisciplinary coordination across the lifespan. At the same time, the therapeutic landscape is rapidly evolving, with cell and gene therapies poised for unprecedented expansion—projected to exceed 200 FDA approvals and result in more than 100,000 patients in the United States receiving these treatments by 2030 (2). Despite this rapid acceleration, many neurologists remain unfamiliar with the genetic testing and patient-selection considerations of emerging disease-modifying therapies (3, 4). Rett syndrome (RTT)—one of the most recognizable monogenic neurodevelopmental disorders—offers a timely and clinically relevant example to illustrate how gene-directed approaches are reshaping expectations for treatment in neurology. This severe X-linked dominant neurodevelopmental disorder, affecting approximately 1 in 10,000–15,000 females, follows a characteristic and devastating trajectory: after a period of apparently normal early development, affected girls experience profound regression of motor and communication skills between 6 and 18 months of age, followed by stabilization with persistent severe impairment (5–8). The disorder is caused by loss-of-function mutations in the MECP2 gene located on chromosome Xq28, which encodes methyl-CpG-binding protein 2 (MeCP2)—a transcriptional regulator essential for neuronal maturation, synaptic plasticity, and the maintenance of mature neuronal function (9, 10). Multiple sophisticated gene therapy strategies for RTT are now in advanced clinical trials, with several more in preclinical and discovery phases, yet a focused review for neurologists is lacking. This article provides a comprehensive, clinician-oriented overview of these emerging therapies, their scientific rationale, current clinical trial data, and the key challenges that remain.
Clinical features, natural history, diagnostic criteria, surveillance, and symptomatic management
2
Understanding the natural history of RTT is essential for interpreting therapeutic trial outcomes and counseling families about realistic expectations. The disorder follows a characteristic four-stage progression, though individual variability is substantial and staging may overlap (11) (Table 1). Diagnosis requires clinical criteria supported by molecular confirmation of a pathogenic MECP2 variant (Table 2). RTT is associated with multiple comorbidities, including epilepsy, which affects 70–90% of individuals and is a major contributor to morbidity. Seizure types are diverse—focal seizures, generalized tonic–clonic seizures, atypical absence seizures, and myoclonic seizures are all reported (12). Up to one third of patients have drug-resistant epilepsy, which caregivers consistently identify as one of the greatest barriers to quality of life (13–17). Beyond epilepsy, additional comorbidities are common and require ongoing multidisciplinary care. Prior to the approval of trofinetide, no disease-specific therapy existed, and management focused primarily on surveillance and symptomatic treatment (18, 19) (Table 3). However, a major limitation—and a source of considerable frustration—is that none of these interventions are truly disease-specific or disease-modifying (20).
Trofinetide
3
Trofinetide, the first FDA-approved therapy for RTT, is a synthetic IGF-1–derived peptide designed to support synaptic function in MECP2-deficient neurons (21–23). Its approval followed a Phase 3 trial demonstrating modest but meaningful clinical improvement, with gastrointestinal side effects as the main tolerability concern (24–28). Open-label extension studies have shown consistent efficacy and no new safety signals (29–32) (Table 4). While trofinetide marks an important milestone and offers symptomatic benefit for some individuals, it requires chronic twice-daily dosing and does not address the underlying MECP2 deficiency—highlighting the need for more transformative, disease-modifying approaches.
With the growing interest in rare disease–specific therapies, numerous pharmacologic treatments for RTT are currently in clinical and preclinical development (summarized in Table 5). However, this review specifically focuses on current and emerging gene therapy approaches.
Gene therapies
4
Since the landmark discovery that restoring MeCP2 expression can reverse severe neurological symptoms — even in mature mice — there has been sustained optimism that gene therapy could meaningfully treat RTT (33). Especially, delivering a functional MECP2 gene to affected brain cells has long been considered a promising therapeutic strategy (34, 35). However, translating this preclinical promise to clinical reality faces a formidable challenge unique to RTT: the biology of X-chromosome inactivation. In females, one X chromosome is randomly inactivated in each cell during early development (lyonization), creating a mosaic pattern where approximately half of neurons express the mutant MECP2 allele while the other half express the wild-type copy (36–40). Conventional gene replacement strategies risk delivering MECP2 to cells already expressing the functional allele, potentially causing overexpression toxicity that mimics MECP2 duplication syndrome—a distinct disorder characterized by intellectual disability, autism, and epilepsy (41). Conversely, insufficient expression in MECP2-deficient neurons fails to achieve therapeutic benefit (42).
Gene replacement therapy for RTT has progressed slowly in mouse models because of multiple challenges: lentiviral vectors provide only limited distribution around the injection site; different generations of expression cassettes using various promoters have carried risks of systemic toxicity—particularly to the liver; transduction efficiency has often been insufficient to rescue phenotypes; newer capsid variants aim to improve blood–brain barrier permeability but may require immunosuppression; and although self-complementary AAVs offer higher efficiency, their reduced packaging capacity (≈2.2 kb) requires careful selection of essential transgene elements (43–50). Consequently, effective gene therapy for RTT demands sophisticated regulatory systems that enable self-tuning, context-dependent MECP2 expression—restoring levels in deficient cells while avoiding toxicity in wild-type cells. The most advanced strategies that have reached clinical trials are discussed below.
Gene replacement therapies: TSHA-102 and NGN-401
4.1
Two gene therapy candidates employing distinct regulatory strategies have entered clinical trials: TSHA-102 (Taysha Gene Therapies) and NGN-401 (Neurogene Inc.) (51). Both use AAV9 vectors for CNS delivery but differ fundamentally in their MECP2 construct design, regulatory mechanisms, and administration routes.
TSHA-102: miniMECP2 with miRARE technology
4.1.1
Molecular design and preclinical foundation
4.1.1.1
TSHA-102 employs a self-complementary AAV9 (scAAV9) vector encoding a truncated “miniMECP2” transgene regulated by the proprietary miRARE (microRNA-Responsive Autoregulatory Element) platform (Figure 1). The miniMECP2 construct addresses a fundamental packaging constraint: scAAV vectors permit faster, more robust gene expression by forming double-stranded DNA without requiring cellular DNA synthesis, but they have a limited cargo capacity of only ~2.2 kilobases. Because the full-length MECP2 coding sequence exceeds this limit, a minimal functional version was engineered that retains only the essential domains: the methyl-CpG-binding domain (MBD), which recognizes methylated DNA, and the NCoR-interaction domain (NID), which recruits transcriptional repressor complexes (52). By delivering the essential MBD and NID functional domains, TSHA-102 provides a genotype-agnostic approach that encompasses the molecular defects found in over 95% of the RTT population, including the eight most common recurrent mutations and nearly all pathogenic missense variants.
TSHA-102 molecular design, miRARE regulation, and preclinical efficacy. (A) Molecular design: TSHA-102 delivers a truncated “miniMECP2” via a self-complementary AAV9 vector, retaining the essential MBD and NID domains to fit the ~2.2 kb scAAV cargo limit. (B) miRARE regulatory mechanism: The miRARE platform inserts microRNA-responsive sequences in the 3’UTR, creating a negative feedback loop: cells with sufficient MeCP2 suppress transgene expression, while MeCP2-deficient cells allow robust expression. (C) Preclinical efficacy: In MECP2-null mice, scAAV-miniMECP2 improved neurological function, breathing, and survival without overexpression toxicity. In non-human primates, lumbar intrathecal delivery achieved broad CNS distribution.
Preclinical validation of miniMECP2 was conducted by delivering scAAV-miniMECP2 directly into the brains of neonatal Mecp2-null mice (52, 53). Treated animals demonstrated significant improvements in neurological function, normalized breathing patterns, and extended survival without the toxicity associated with full-length MECP2 overexpression in wild-type mice (52). This established proof-of-concept that a truncated MECP2 protein, if efficiently expressed, could rescue core RTT phenotypes.
However, even miniMECP2 poses risks of overexpression. To address this, the miRARE regulatory system was utilized (54). miRARE leverages endogenous microRNAs to create a negative feedback loop: multiple tandem sequences complementary to specific endogenous miRNAs are inserted into the 3′ untranslated region (3’UTR) of the miniMECP2 transgene. In cells where these cognate miRNAs are abundant (including neurons that already express sufficient MeCP2), miRNA binding recruits the RNA-induced silencing complex (RISC), which degrades the transgene mRNA or blocks its translation. In MECP2-deficient cells, where these regulatory miRNAs are expressed differently, the transgene is expressed more robustly.
The elegance of miRARE lies in its self-tuning behavior. In preclinical studies, AAV9/miniMECP2-miRARE restricted expression to only ~8% of neurons in wild-type mice, preventing overexpression toxicity (54). But in Mecp2-null mice, expression expanded to ~40% of neurons—sufficient to produce therapeutic benefit (54). Treated knockout mice showed delayed onset of motor abnormalities, improved gait, normalized breathing, and significantly prolonged survival. Importantly, the system appeared to adapt expression levels to cellular need, with cells already expressing adequate MeCP2 suppressing the transgene while deficient cells permitted robust expression. In non-human primates, lumbar intrathecal administration resulted in broad and uniform biodistribution throughout the brain and spinal cord, comparable to that achieved with intracisternal magna delivery (55).
Clinical development: the REVEAL trials
4.1.1.2
miniMECP2 with miRARE technology is being further investigated, with the REVEAL clinical development program comprising two Phase 1/2/3 studies (NCT06152237 and NCT05606614). Both are open-label, multicenter trials with Part A dose-escalation and Part B dose-expansion components designed to evaluate the safety, tolerability, and preliminary efficacy of intrathecally administered TSHA-102 (56, 57).
REVEAL pediatric study — eligibility and treatment
4.1.1.3
The REVEAL Pediatric Study enrolled females aged 5–8 years with genetically confirmed classical RTT due to pathogenic MECP2 variants, a Clinical Global Impression–Severity (CGI-S) score ≥4, and no need for mechanical ventilation. Participants received a single intrathecal bolus via lumbar puncture at either 5.7 × 10^14^ or 1 × 10^15^ vector genomes. Immunosuppression, typically corticosteroids, begins 7 days before dosing to mitigate immune responses to the AAV vector. Post-treatment monitoring extends up to 6 years.
Primary endpoints focus on safety—incidence and severity of adverse events (AEs), serious AEs, and events of special interest including immune-mediated reactions, hepatotoxicity, neurologic worsening, and seizure exacerbation. Secondary endpoints evaluate preliminary efficacy using validated and exploratory measures including CGI-I/CGI-S scales, the Revised Motor Behavior Assessment (R-MBA), the Mullen Scales of Early Learning adapted for RTT (MSEL-A), Observer-Reported Communication Ability (ORCA), quantitative EEG metrics (auditory/visual evoked potentials), and monthly seizure frequency.
Part A results: safety and preliminary efficacy
4.1.1.4
Topline Part A results released in 2024 demonstrated a favorable safety profile. TSHA-102 was well tolerated at both dose levels, with no treatment-related SAEs or dose-limiting toxicities (58). Most treatment-emergent AEs were mild to moderate and included transient elevations of liver transaminases (AST/ALT), pyrexia, and lethargy. Transaminase elevations were generally asymptomatic and responsive to steroid therapy. Temporary increases in cerebrospinal fluid neurofilament light chain (NfL) were observed in some participants but were not associated with neurologic decline. Notably, seizure control was maintained or improved following TSHA-102 administration. Enrollment of six participants in Part A is complete.
Preliminary efficacy exceeded natural-history expectations. All six participants achieved meaningful functional gains, with each acquiring or regaining at least one developmental milestone considered highly unlikely to occur spontaneously based on published natural-history data (<6.7% milestone acquisition in girls ≥6 years) (Table 6). Improvements spanned communication, fine-motor, and gross-motor domains—for example, responding to familiar names, following commands, using pincer grasp., self-feeding, pulling to stand, walking independently, and climbing stairs (59). Improvements emerged within 3–6 months and were sustained with continued follow-up. Standardized scale-based outcomes (MSEL-A, R-MBA, ORCA) corroborated these findings, and blinded centralized review of video evidence confirmed milestone achievements.
A clear dose–response relationship was observed: participants receiving 1 × 10^15^ vg demonstrated faster and more pronounced functional gains than those receiving 5.7 × 10^14^ vg. This supports biological plausibility and informed dose selection for Part B.
Part B (pivotal expansion) — ongoing
4.1.1.5
The ongoing REVEAL pivotal expansion trial (NCT05606614) will enroll 15 females aged 6 to <22 years with typical RTT to further characterize safety and efficacy in a broader, more heterogeneous population. As of the May 2025 data cut, 12 participants (low dose n = 4; high dose n = 8) had received TSHA-102, representing a diverse range of MECP2 variants and clinical severities (60).
Safety outcomes remain consistent with Part A (60). Efficacy data (N = 10) reflect cumulative functional improvement (60). Participants achieved a total of 22 developmental milestones, as verified through independent, blinded video adjudication using prespecified milestone definitions. In addition, 165 incremental skill gains were recorded across validated scales. At ≥9 months post-treatment, high-dose recipients consistently outperformed the low-dose cohort across multiple domains, demonstrating deepening dose-dependent benefit over time. Mean R-MBA score improved by −11.5 points in the low-dose cohort and −18.0 in the high-dose cohort. Mean CGI-I scores at ≥9 months were 2.8 and 1.0 for low- and high-dose cohorts, respectively. CGI-S improvement was recorded in 25% of low-dose and 33% of high-dose recipients at latest follow-up.
NGN-401: full-length MECP2 with EXACT technology
4.1.2
Molecular design and preclinical foundation
4.1.2.1
NGN-401 (Neurogene Inc.) takes a conceptually distinct approach: it delivers the full-length human MECP2 gene rather than a truncated version, employing a novel self-regulating circuit called EXACT (Expression Attenuation via Construct Tuning) to control expression levels (Figure 2).
EXACT-mediated regulation of MECP2 transgene expression. The MECP2 transgene (blue) is transcribed into mRNA (blue lines) containing EXACT1 microRNA binding sites in the 3’UTR (pale blue symbols). A portion of the transcripts is translated into full-length MeCP2 protein (right). To prevent overexpression, EXACT1 microRNA (yellow/orange triangles) is co-expressed in proportion to the transgene. EXACT1 binds to its target sites on MECP2 transcripts, selectively degrading excess mRNA (left), thereby maintaining physiologic MeCP2 protein levels.
The EXACT system implements a synthetic, cell-autonomous, incoherent feedforward loop (61). A single ubiquitous promoter drives transcription of both the MECP2 coding sequence and a designed, non-mammalian microRNA (EXACT1) (61). EXACT1 is engineered to recognize complementary target sites inserted into the 3′UTR of the same MECP2 transcript, creating an intrinsic negative feedback mechanism. When MECP2 expression rises, increased production of EXACT1 microRNA suppresses MECP2 translation; when expression falls, reduced EXACT1 levels relieve this repression. This design produces a dose-independent, self-buffering circuit that stabilizes cellular MECP2 abundance regardless of viral genome copy number.
Preclinical validation demonstrated the utility of this approach. In cell-based assays, EXACT-regulated constructs exhibited a significantly narrower distribution of MeCP2 protein levels across cells compared to conventional unregulated constructs (61). Importantly, the degree of regulation increased at higher transgene doses—precisely when the risk of overexpression toxicity is greatest. In Mecp2−/y hemizygous male mice, intracerebroventricular (ICV) administration of NGN-401 at postnatal day 1–2 resulted in prolonged survival (from ~10 weeks to >1 year) and amelioration of RTT-like motor, respiratory, and behavioral phenotypes (61). Critically, NGN-401 was well tolerated in female Mecp2+/− mice (the model more relevant to human RTT given mosaicism) and healthy juvenile non-human primates without causing the toxicity seen with unregulated MECP2 overexpression constructs (61).
Route of administration: ICV vs. intrathecal
4.1.2.2
A significant differentiator of NGN-401 is its intended route of administration: ICV injection rather than intrathecal lumbar puncture. Recent preclinical studies in nonhuman primates (juvenile cynomolgus macaques) compared ICV versus intrathecal lumbar (IT-L) delivery. ICV administration achieved markedly superior AAV biodistribution throughout the brain, with 10–100 × higher transgene RNA expression in cortical regions, deep brain structures, and upper spinal cord compared to equivalent or even higher IT-L doses (62). Importantly, systemic (liver) exposure was comparable between routes, indicating no liver-sparing advantage of IT-L delivery and suggesting similar systemic biodistribution despite the different injection sites (62). ICV delivery requires neurosurgical placement of a catheter into the lateral ventricle—a more invasive procedure than lumbar puncture but one routinely performed for ventriculoperitoneal shunt placement in pediatric neurosurgery centers. The improved CNS biodistribution may translate to enhanced efficacy, but it also necessitates greater neurosurgical expertise and infrastructure.
Clinical development: the EMBOLDEN trial
4.1.2.3
NGN-401 is being evaluated in the EMBOLDEN (previously ASPIRE) trial (NCT05898620), initially designed as a Phase 1/2 dose-escalation study that has now transitioned to a pivotal, open-label, baseline-controlled, multicenter, single-arm efficacy trial.
Eligibility criteria include females with genetically confirmed typical RTT and a documented pathogenic MECP2 mutation, stable antiseizure medication regimen for ≥12 weeks, post-regression stage, and either trofinetide-naïve status or prior discontinuation of trofinetide. The trial originally enrolled participants into sequential arms by age: Arms 1 and 2 (females aged 4–10 years) and Arm 3 (females ≥11 years) tested escalating doses. Arm 4, the pivotal cohort, enrolls females aged ≥3 years.
The primary efficacy endpoint is the proportion of responders defined by: (1) achieving a Clinical Global Impression-Improvement (CGI-I) score ≤3 (“minimally improved” or better) AND (2) gaining at least one developmental milestone from a predefined list of 28 skills across fine motor, gross motor, and communication/social domains, assessed via standardized video recordings independently reviewed by blinded central raters over 52 weeks. This composite endpoint ensures that both objective skill acquisition and global clinical meaningfulness are captured. A total of 33 participants will be evaluated for efficacy and safety over a three-year post-treatment period, with planned enrollment in a subsequent 12-year long-term follow-up study.
Early clinical results: efficacy and safety signals
4.1.2.4
Early results from the low-dose cohort (1 × 10^15^ vg) demonstrated a favorable safety profile and encouraging efficacy (63). The first four treated girls achieved meaningful functional gains, acquiring 23 developmental skills collectively. Improvements spanned all functional categories: fine motor (reaching, grasping, finger feeding, utensil use), gross motor (sitting, standing, walking, climbing stairs), and communication (responding to names, following commands, pointing, waving, babbling, using words with meaning).
These gains, which would be exceedingly rare based on natural history (<6.7% spontaneous milestone acquisition rate), occurred early post-treatment and were sustained through ongoing follow-up. Clinician and caregiver assessments consistently rated participants as improved, supporting the clinical meaningfulness of observed changes.
The high-dose safety event
4.1.2.5
A participant treated with the high dose (3 × 10^15^ vg) developed systemic hyperinflammatory syndrome several days post-dosing, characterized by fever, cytopenia, markedly elevated ferritin, and multi-organ dysfunction consistent with hemophagocytic lymphohistiocytosis (HLH) or macrophage activation syndrome (MAS) (64). Despite aggressive treatment with corticosteroids and immunosuppressive therapy, the participant succumbed to complications of the inflammatory syndrome. Two other participants had previously received the same 3 × 10^15^ vg dose without developing hyperinflammatory reactions, suggesting that individual susceptibility factors—such as subclinical infection, immune priming, or a genetic predisposition to HLH—may have contributed, rather than dose-dependent toxicity alone. However, high-dose treatments were immediately discontinued, and the maximum allowable dose was capped at 1 × 10^14^ vg/kg, with no participant receiving more than 1 × 10^15^ vg in total. Additional safety measures were implemented to reduce the risk of hyperinflammatory complications. Infection screening was strengthened to exclude any systemic illness within 30 days of dosing, including testing for EBV and CMV, and to rule out COVID-19 within 6 weeks. Before dosing, sites are now required to have anakinra (an IL-1 receptor antagonist and first-line therapy for HLH/MAS) immediately available and to coordinate with local HLH specialists. Post-dosing surveillance was intensified with daily assessments during the first week, including ferritin, fever, and blood counts to monitor for the classical HLH triad. A stepwise treatment algorithm was added, beginning with high-dose corticosteroids and escalating to anakinra for refractory cases. HLH-specific treatment pathways were also formally integrated into the protocol.
Both TSHA-102 and NGN-401 represent scientifically sophisticated attempts to overcome the unique challenges of RTT gene therapy, but they differ in key aspects (Table 7). Both programs have demonstrated proof-of-concept efficacy—functional gains across multiple domains that far exceed natural history expectations. The critical questions moving forward include: (1) whether full-length MECP2 (NGN-401) offers advantages over miniMECP2 (TSHA-102) in terms of functional protein activity; (2) whether ICV delivery’s superior biodistribution translates to greater clinical efficacy; (3) whether the enhanced safety protocols for NGN-401 adequately mitigate hyperinflammatory risk; and (4) what the optimal timing and patient selection criteria are for each approach.
Emerging and next-generation approaches
5
Beyond AAV-mediated gene replacement, several innovative therapeutic strategies are progressing through preclinical development, each offering distinct advantages and facing unique challenges (Figure 3).
Innovative genetic approaches being explored for Rett syndrome: (A) gene editing using CRISPR-Cas9, which enables either broad modifications in DNA segments or precise single-nucleotide changes. (B) RNA editing via the ADAR system, which converts adenosine into inosine through deamination; inosine is interpreted as guanosine and pairs with cytosine in the tRNA anticodon. (C) Left panel: a premature termination codon (PTC) halts mRNA translation. Right panel: a specialized suppressor tRNA binds the PTC, permitting translation to continue. Erf1 refers to eukaryotic translation termination factor 1. (D) The mutant MECP2 allele (red M) resides on the active X chromosome (Xa), while the wild-type allele (black W) is on the inactive X chromosome (Xi). Reactivation of the wild-type allele (Xi) can be achieved through chemical inhibition of silencing factors or epigenetic modulation, such as blocking DNA methylation. Reproduced from Palmieri et al. (95), under a Creative Commons Attribution License (CC BY 4.0).*
CRISPR-Cas9 genome editing
5.1
CRISPR-Cas9 genome editing offers the advantage of directly repairing pathogenic MECP2 mutations at the DNA level, providing the potential for permanent correction without the need for continuous transgene expression (65). The system uses a guide RNA to direct the Cas9 nuclease to a specific genomic site, where a double-strand break is introduced and subsequently repaired by non-homologous end joining or homology-directed repair using a supplied template (66).
Proof-of-concept work has shown successful correction of MECP2 mutations in patient-derived induced pluripotent stem cells (iPSCs) with repair of mutation in high efficiency (67, 68). Clinical translation is being pursued through the MECPer-3D program (NCT05740761), which focuses on CRISPR-Cas9 correction of the four most common MECP2 hotspot mutations—c.473C > T (p.T158M), c.502C > T (p.R168X), c.763C > T (p.R255X), and c.916C > T (p.R306C). The program includes development of mutation-specific guide RNAs and repair templates, evaluation of editing efficiency and specificity in patient-derived cellular models, assessment of off-target events through whole-genome sequencing, and preclinical testing in mouse and potentially large-animal models. Key endpoints include the proportion of alleles successfully edited and the characterization of any off-target genomic changes.
Several barriers remain for clinical application, including effective delivery of CRISPR components across the central nervous system, achieving sufficient editing efficiency to translate into meaningful clinical benefit, minimizing off-target effects, and ensuring safe dosage given the narrow therapeutic window of MeCP2 and the irreversible nature of genomic editing. Mosaic correction is also expected, as only transduced cells will be edited; however, even a shift toward a greater proportion of functional neurons may be beneficial. Despite these challenges, genome editing represents a promising long-term strategy because of its potential as a one-time, durable corrective treatment.
RNA editing with ADAR
5.2
An alternative to permanent DNA editing is transient correction at the RNA level using adenosine deaminases acting on RNA (ADARs). ADARs catalyze adenosine-to-inosine (A → I) editing in double-stranded RNA, and because inosine is interpreted as guanosine (G) during translation, this strategy enables correction of specific point mutations—including pathogenic G > A variants—directly at the mRNA level. Sinnamon et al. provided proof-of-concept for MECP2 RNA editing in mouse neurons by engineering an ADAR2 catalytic domain fused to an RNA-binding protein that guides editing to Mecp2 mRNA (69). This approach produced up to 72% editing efficiency, restored MeCP2 protein levels and heterochromatin localization, and improved electrophysiological properties and gene-expression patterns in edited neurons (69, 70).
RNA editing offers several advantages: its effects are reversible and tunable, requiring repeated dosing similar to ASOs; it poses a lower risk of off-target consequences compared with permanent DNA modification; and in principle it can treat multiple mutation types by redirecting ADAR activity to different transcript sites. However, key challenges remain, including achieving sufficient editing efficiency across enough neurons, optimizing CNS delivery for either repeated dosing or long-term AAV-mediated ADAR expression, controlling immunogenicity of engineered ADAR proteins, ensuring high target specificity, and addressing durability, as ongoing administration or sustained transgene expression is required. Although still in early preclinical development, RNA editing represents a promising middle ground between symptomatic treatment and irreversible genome editing, offering the potential to correct pathogenic MECP2 mutations without altering DNA permanently.
Translation readthrough for nonsense mutations
5.3
Approximately 30–35% of pathogenic MECP2 variants are nonsense mutations that prematurely truncate the protein (71). Translation readthrough strategies aim to suppress these premature termination codons (PTCs), allowing ribosomes to produce near–full-length MeCP2. Two approaches are under investigation: small-molecule readthrough compounds, including aminoglycosides (e.g., gentamicin, G418) and designer drugs like PTC124 (ataluren), which alter ribosomal fidelity to incorporate an amino acid at the stop codon; and suppressor tRNAs engineered to recognize specific stop codons and insert an amino acid to enable continued translation (71–73).
While these strategies have shown promise in other genetic disorders such as Duchenne muscular dystrophy and cystic fibrosis, their adaptation to RTT faces significant challenges. Readthrough must be efficient enough to restore functional MeCP2 but selective enough to avoid suppressing normal stop codons across the genome, which could create off-target protein extensions. Moreover, the amino acid incorporated at the PTC may differ from the wild-type residue, potentially impacting protein function. MeCP2’s narrow dosage sensitivity further complicates translation readthrough, as even successful suppression must achieve expression within the therapeutic window. Translation readthrough for RTT remains in early discovery, with proof-of-concept studies ongoing in cellular models, and no clinical programs have yet been initiated.
X-chromosome reactivation: unlocking the endogenous wild-type allele
5.4
Perhaps the most conceptually elegant approach for female RTT patients is reactivation of the silenced wild-type MECP2 allele on the inactive X chromosome (Xi) (74–76). Because females undergo random X-inactivation early in development, approximately half of neurons express the mutant allele while the remaining half carry a functional, but epigenetically silenced, MECP2 copy. Selectively reactivating this Xi-linked wild-type allele—without globally disrupting X-chromosome dosage compensation—could restore physiological MeCP2 levels using the patient’s own endogenous gene.
Earlier work also demonstrated that combined inhibition of ACVR1 and PDPK1, two regulators of X-chromosome silencing, can reactivate Xi-linked Mecp2 in neurons and achieve partial in vivo reactivation in heterozygous female mouse brains (77, 78). Large-scale functional screens have identified multiple regulatory pathways that control MECP2 silencing on the inactive X chromosome (Xi). An RNAi screen in RTT patient–derived cells revealed that components of the BMP/TGF-β signaling pathway modulate expression of XIST, the long noncoding RNA that governs X-inactivation; inhibiting this pathway partially reactivated Xi-linked MECP2 without inducing global X-chromosome reactivation (79). Follow-up studies showed that small-molecule BMP inhibitors can similarly restore MECP2 expression in primary neurons from Mecp2-heterozygous female mice, providing pharmacologic proof-of-concept (78).
Another study demonstrated that pairing an antisense oligonucleotide (ASO) targeting Xist RNA with the DNA methylation inhibitor 5-azacytidine (5-Aza) produced robust reactivation of a silent Mecp2 luciferase reporter in mouse fibroblasts, highlighting the need to simultaneously disrupt RNA-based and DNA-based silencing layers (75). High-throughput screening also identified JAK/STAT pathway inhibitors (AG490, Jaki) as additional modulators of Xi stability, although reactivation effects were strongly cell-type dependent—effective in mouse fibroblasts but limited in human Xi-containing cell lines (80).
More recently, a genome-wide CRISPR loss-of-function screen in female fibroblasts identified miR-106a as a key regulator of the RNA-based silencing machinery that stabilizes Xi (81). Inhibiting miR-106a disrupted this system, destabilized the inactive X chromosome, and produced therapeutic benefits in RTT mouse models—extending survival, improving motor and exploratory behaviors, and reducing breathing abnormalities (81).
To achieve gene-specific reactivation without globally perturbing X-chromosome dosage, targeted epigenetic editing has emerged as a promising strategy. One study used dCas9–Tet1–mediated promoter demethylation to selectively reactivate the Xi-linked MECP2 allele in human embryonic stem cell–derived neurons, achieving up to 82% restoration of MeCP2 protein with minimal off-target effects (82). Similar approaches reactivated Xi-linked MECP2-GFP in neurons at more modest levels (~18%) but confirmed allele specificity without widespread reactivation of other X-linked genes (82). However, the large size of dCas9–Tet1 poses significant challenges for AAV-mediated CNS delivery in vivo.
Together, these studies demonstrate that X-chromosome reactivation represents a compelling therapeutic avenue capable of restoring endogenous MECP2 expression, avoiding the immunogenicity and dosage risks of gene-addition therapies, and potentially enabling small-molecule or ASO-based clinical approaches. Key challenges remain—including achieving brain-wide and allele-specific reactivation, ensuring CNS penetrance of candidate therapies, avoiding unintended activation of other X-linked genes, and determining whether continuous treatment will be required. Nonetheless, ongoing dissection of the druggable regulatory nodes governing Xi maintenance continues to advance this strategy toward translational feasibility.
Critical considerations for clinical translation
6
Optimal therapeutic window and timing of intervention
6.1
A central question for clinical translation is when gene therapy should be delivered. Mouse studies show that restoring MeCP2 can be effective across a wide age range—from presymptomatic neonates to fully symptomatic adults—indicating that the Rett phenotype remains at least partly reversible even after prolonged deficiency. Nonetheless, several considerations favor earlier intervention: the first 2–3 years of life represent critical windows of synaptogenesis and circuit formation; prolonged MeCP2 deficiency may lead to structural neuronal and glial changes that become harder to reverse; and treating before or during early regression could prevent milestone loss rather than requiring later recovery (83, 84). Conversely, treatment of older individuals has advantages including greater diagnostic certainty, a clearer risk–benefit rationale in severely affected patients, and more straightforward outcome assessment. Current clinical trials enroll patients ages 3–20+, reflecting uncertainty about the optimal window. As safety becomes better defined, intervention in younger children—including presymptomatic infants identified by genetic screening—will likely be explored. Ultimately, a stratified approach may emerge in which early treatment maximizes developmental potential while older individuals are treated to achieve meaningful functional gains. In summary, while pharmacological agents like Trofinetide are effective across a broad age range (2 years to adult), emerging gene therapies such as TSHA-102 and NGN-401 (currently recruiting down to age 3 in pivotal trials) suggest a potential for functional ‘regain’ that is highly sensitive to the developmental window of administration.
Dosing, biodistribution, and the “enough but not too much” problem
6.2
Gene therapy for RTT requires dosing that is both effective and safe, balancing transduction thresholds with the risk of overexpression toxicity. It remains unclear what percentage of brain cells must express the transgene in humans, whether certain regions (e.g., cortex, brainstem) require preferential targeting, and how neuronal versus glial transduction contributes to benefit (83, 85). The fatal hyperinflammatory reaction observed with high-dose NGN-401 highlights that toxicity may arise not only from excessive MeCP2 but also from high systemic AAV loads. Administration route strongly influences biodistribution: ICV delivery (NGN-401) provides broad brain penetration but requires neurosurgery, whereas intrathecal lumbar injection (TSHA-102) is less invasive but may yield more limited forebrain distribution. Future approaches—such as optimized injection routes, convection-enhanced delivery, nanoparticle-based gene delivery systems, or next-generation AAV capsids with improved CNS tropism—may help navigate this therapeutic window (86, 87).
Measuring meaningful change
6.3
Efficacy assessment in RTT trials is challenging due to floor effects on standard developmental tests, lack of validated disease-specific tools, and the risk of bias in caregiver-reported outcomes. Recently developed milestone checklists address disease specificity but still require ongoing validation and regulatory acceptance. Video-based blinded raters improve objectivity but increase logistical complexity. Determining what constitutes clinically meaningful benefit is equally difficult; families report that even modest gains in independence, communication, or daily living skills are profoundly valuable. Natural history variability further complicates interpretation of single-arm studies, necessitating robust genotype-informed natural history datasets. Current trials use composite endpoints that combine milestone achievement with CGI-I scores, while multiple candidate biomarkers (EEG signatures, neuroimaging connectivity, eye-tracking, and brain-derived plasma microRNAs) may ultimately provide objective surrogate endpoints if validated against functional outcomes (88–94).
Long-term safety
6.4
Because gene therapy aims to deliver lifelong MeCP2 expression, long-term safety remains largely unknown. Open questions include durability of expression, feasibility of re-dosing in the context of anti-AAV immunity, potential for delayed toxicity from MeCP2 overexpression, risk of chronic CNS immune responses, and the theoretical possibility of insertional mutagenesis despite low AAV integration rates. Another consideration is whether restoring MeCP2 in a brain that developed under deficiency could introduce new vulnerabilities. Addressing these uncertainties will require decades-long surveillance; current protocols mandate 15-year follow-up for all treated individuals.
Access, equity, and economic barriers
6.5
Gene therapy for RTT is expected to cost in the range of other approved AAV therapies ($1–3 M+), raising questions about payer coverage even if treatment reduces long-term care costs. Access is further constrained by the need for specialized centers capable of administering therapy and monitoring for complications, disproportionately affecting families in rural areas, underserved communities, and low-income countries. Regulatory and reimbursement heterogeneity across regions will further influence global availability. Patient advocacy organizations have played a central role in advancing research, but ensuring equitable access after approval will require policy coordination, financing innovation, and commitment from manufacturers and healthcare systems.
Ethical considerations
6.6
Gene therapy trials in severely affected children raise complex ethical questions. Families must make decisions amid profound emotional pressure, uncertain benefit, and real risks—including catastrophic adverse events. Clinicians and researchers must communicate risks and uncertainties honestly, avoiding both pessimism that discourages participation and optimism that overstates benefit. Early-phase studies blur the boundary between research and therapy, especially when early improvement fuels calls for expanded access. Equity concerns also arise if transformative treatments ultimately reach only children with geographic or financial privilege.
Conclusion
7
RTT highlights both the promise and the complexity of precision medicine for neurodevelopmental disorders. In just over two decades, the field has moved from identifying MECP2 as the causative gene to demonstrating phenotypic reversibility in animal models and now to evaluating multiple gene therapies in human trials. Trofinetide marked the first disease-specific therapy, while gene-replacement approaches such as TSHA-102 and NGN-401 hold the potential for far greater clinical impact and have already shown early signals of meaningful functional improvement in treated children. At the same time, enthusiasm must be balanced with caution. Serious adverse events, uncertainties surrounding optimal dosing, timing, durability, and long-term safety, and persistent barriers to access and affordability underscore the need for rigorous science and responsible translation. As these therapies advance, neurologists will play a central role in guiding families, supporting clinical trials, preparing systems for delivery, and advocating for equitable access. The coming years will determine whether decades of discovery culminate in transformative, widely accessible treatment for individuals with RTT. Achieving that goal will require continued innovation, sustained vigilance, and a commitment to ensuring that every affected individual has the opportunity to benefit from emerging therapeutic breakthroughs.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Nguengang Wakap S Lambert DM Olry A Rodwell C Gueydan C Lanneau V . Estimating cumulative point prevalence of rare diseases: analysis of the Orphanet database. Eur J Hum Genet. (2020) 28:165–73. doi: 10.1038/s 41431-019-0508-031527858 PMC 6974615 · doi ↗ · pubmed ↗
- 2Cardinalhealth. 2025. Advanced therapies report. Available online at: https://www.cardinalhealth.com/En/Services/Manufacturer/Biopharmaceutical/Cell-and-Gene-Therapies/Advanced-Therapies-Report.Html. [Accessed October 12, 2025].
- 3Jaitovich Groisman I Hurlimann T Shoham A Godard B. Practices and views of neurologists regarding the use of whole-genome sequencing in clinical settings: a web-based survey. Eur J Hum Genet. (2017) 25:801–8. doi: 10.1038/ejhg.2017.64, 28488681 PMC 5520076 · doi ↗ · pubmed ↗
- 4Salm M Abbate K Appelbaum P Ottman R Chung W Marder K . Use of genetic tests among neurologists and psychiatrists: knowledge, attitudes, behaviors, and needs for training. J Genet Couns. (2014) 23:156–63. doi: 10.1007/s 10897-013-9624-0, 23793969 PMC 3812264 · doi ↗ · pubmed ↗
- 5Petriti U Dudman DC Scosyrev E Lopez-Leon S. Global prevalence of Rett syndrome: systematic review and Meta-analysis. Syst Rev. (2023) 12:5. doi: 10.1186/s 13643-023-02169-6, 36642718 PMC 9841621 · doi ↗ · pubmed ↗
- 6Percy AK Lane JB. Rett syndrome: clinical and molecular update. Curr Opin Pediatr. (2004) 16:670–7. doi: 10.1097/01.mop.0000143693.59408.ce, 15548931 · doi ↗ · pubmed ↗
- 7Rett A. On a unusual brain atrophy syndrome in Hyperammonemia in childhood. Wien Med Wochenschr. (1966) 116:723–6.5300597 · pubmed ↗
- 8Hagberg B Aicardi J Dias K Ramos O. A progressive syndrome of autism, dementia, Ataxia, and loss of purposeful hand use in girls: Rett's syndrome: report of 35 cases. Ann Neurol. (1983) 14:471–9. doi: 10.1002/ana.410140412, 6638958 · doi ↗ · pubmed ↗