ACTA1-related congenital myopathy in a neonate: a case report and literature review
Lingxia Zhao, Feng Deng, Baohuan Cai

TL;DR
This case report describes a rare genetic disorder in a newborn, highlighting the importance of genetic testing and early care planning.
Contribution
The study emphasizes the diagnostic challenges and management of ACTA1-related congenital myopathy in neonates.
Findings
ACTA1-related myopathy can mimic perinatal asphyxia, requiring genetic testing for accurate diagnosis.
Early-onset ACTA1 mutations are associated with severe prognosis and need for early palliative care.
Precise genetic diagnosis is crucial for understanding the condition and developing future therapies.
Abstract
ACTA1-related congenital myopathies are rare neuromuscular disorders with significant genotypic heterogeneity, often causing severe neonatal multisystem involvement. This study presents a severe neonatal case with a pathogenic ACTA1 variant and reviews literature to highlight diagnostic and management challenges. A female infant was born via cesarean section at 39+1 weeks to a non-consanguineous mother. Prenatal ultrasound showed polyhydramnios. She presented with severe birth asphyxia (Apgar 3 at 1 min, 5 at 5 min), requiring immediate resuscitation. Physical examination revealed profound hypotonia, absent spontaneous movements, respiratory insufficiency necessitating mechanical ventilation, expressionless facies, and bulbar dysfunction. Laboratory tests indicated metabolic acidosis and elevated lactate and creatine kinase. Electromyography (EMG) demonstrated reduced motor amplitudes…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
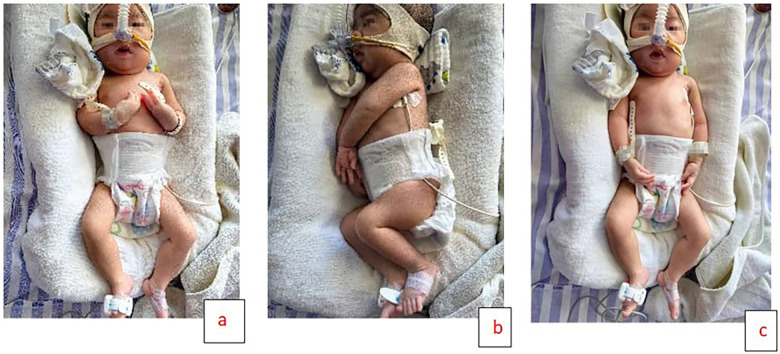 Figure 1
Figure 1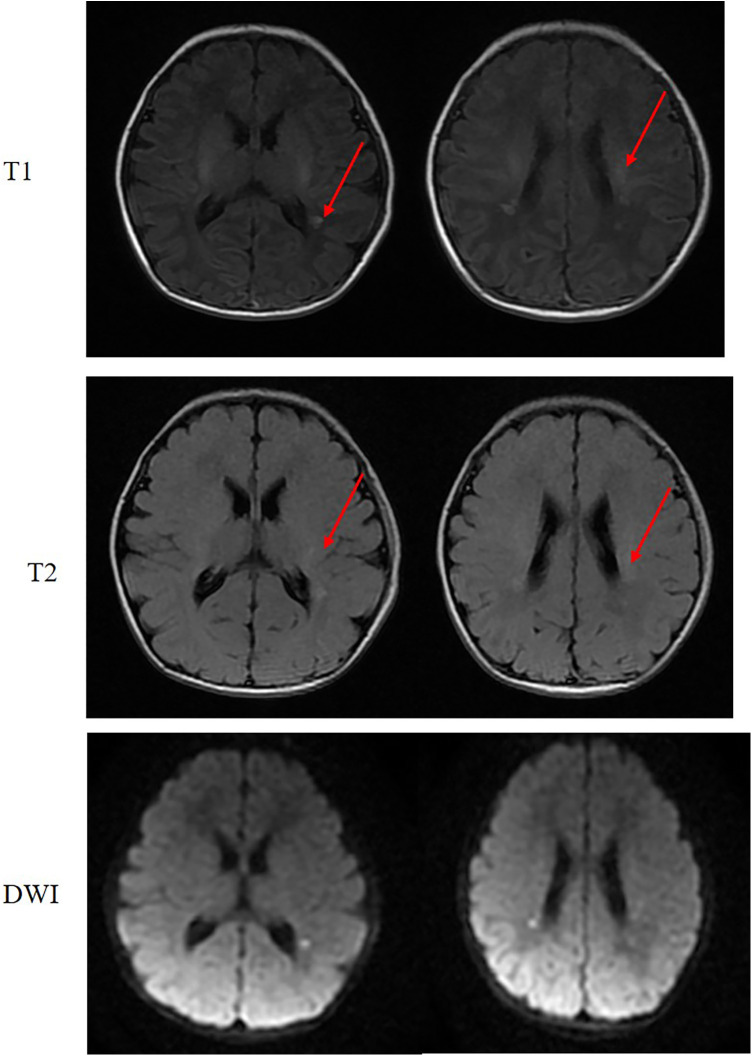 Figure 2
Figure 2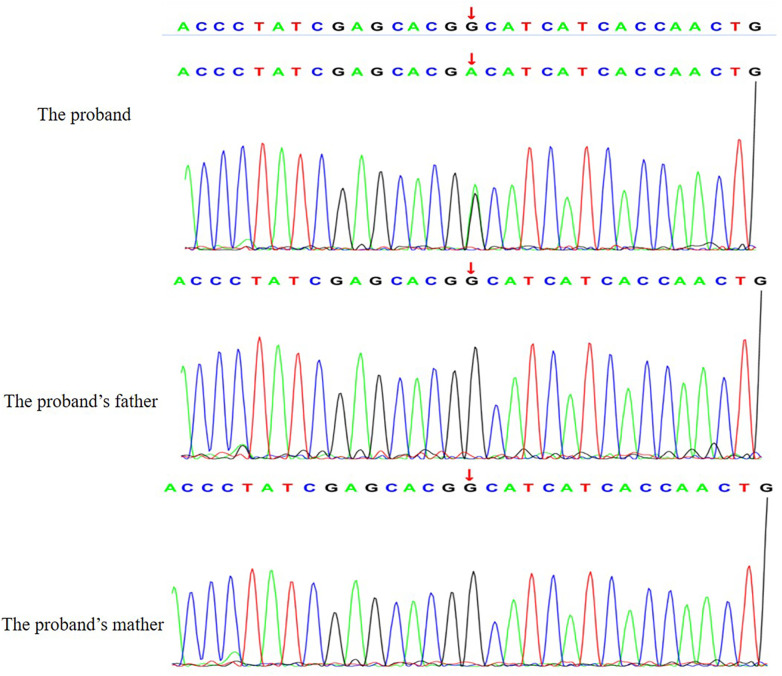 Figure 3
Figure 3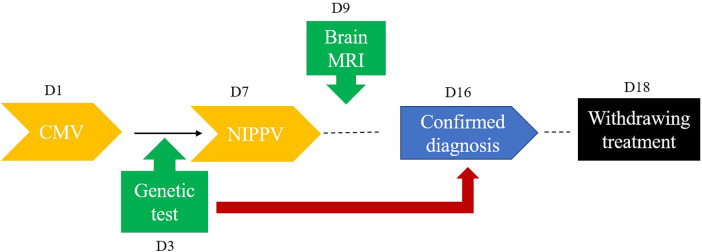 Figure 4
Figure 4- —Natural Science Foundation of Hubei Province10.13039/501100003819
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCardiomyopathy and Myosin Studies · Hereditary Neurological Disorders · Neurogenetic and Muscular Disorders Research
Introduction
Congenital myopathies represent a heterogeneous group of inherited neuromuscular disorders characterized by structural abnormalities in skeletal muscle fibers. They typically present with hypotonia, muscle weakness, and respiratory insufficiency beginning in infancy or early childhood (1). Among the various genetic causes, mutations in the ACTA1 gene, which encodes skeletal muscle α-actin, account for approximately 15%–25% of cases. These mutations are associated with distinct histopathological subtypes, including nemaline myopathy, actin aggregate myopathy, and congenital fiber-type disproportion (CFTD) (2, 3).
Neonatal-onset ACTA1-related myopathy is particularly severe, often manifesting as fetal akinesia, arthrogryposis, polyhydramnios, and immediate respiratory failure at birth (4). Despite advances in genetic diagnostics, the condition is frequently misdiagnosed as hypoxic-ischemic encephalopathy (HIE) or spinal muscular atrophy due to overlapping clinical features (5). This diagnostic delay impedes timely genetic counseling and complicates prognostication and family planning. The prognosis is generally poor, with over 40% of neonatal cases succumbing to respiratory complications within the first year of life (6). Given the severity, phenotypic variability, and challenges in early recognition, there is a continued need to delineate the clinical and genetic spectrum of this disorder.
In this article, we report a severe neonatal case caused by a pathogenic ACTA1 variant, and provide a comprehensive review of previously reported cases to improve awareness, facilitate early diagnosis, and highlight multidisciplinary management challenges.
Case presentation
The proband was a female infant born at 39^+1^ weeks via cesarean section to a 29-year-old G3P2 mother with a history of uterine scarring. Prenatal ultrasound detected polyhydramnios (amniotic fluid volume: 2,500 ml), but no other structural anomalies were identified. There was no consanguinity between parents, and family history was negative for neuromuscular or metabolic disorders. The proband's 7-year-old sibling exhibited normal motor development. At delivery, the infant demonstrated severe birth asphyxia, with Apgar scores of 3 and 5 at 1 and 5 min, respectively, necessitating immediate resuscitation. Upon transfer to the neonatal intensive care unit, she exhibited lethargy, poor responsiveness, and an expressionless facies. Neuromuscular examination revealed profound generalized hypotonia and absence of spontaneous movements, with only minimal finger and toe motion observed. She developed respiratory insufficiency requiring mechanical ventilation. Bulbar dysfunction was prominent, manifesting as weak sucking and swallowing reflexes along with excessive oral secretions. Primitive reflexes including Moro and grasp were absent, although ocular movements remained intact (Figure 1).
Three-panel image of a newborn undergoing medical treatment. Panel a and c show the baby lying on their back with monitoring devices attached. Panel b shows the baby lying on their side with similar equipment.
Initial laboratory studies revealed metabolic acidosis (pH 7.27, pCO₂ 46.2 mmHg, HCO₃⁻ 20 mmol/L, SBE −5.3 mmol/L) with elevated lactate (2.7 mmol/L). Cardiac evaluation demonstrated markedly elevated creatine kinase (peak 578 U/L, normalizing by Day 9), while electrocardiography (ECG) showed sinus tachycardia (177 bpm) with ventricular ectopy. Echocardiographic findings were notable for a hemodynamically insignificant patent ductus arteriosus (1.2 mm) with preserved left ventricular function (ejection fraction 53%). Neurophysiological assessment identified multifocal epileptiform discharges and subclinical seizures on Day 1 electroencephalography (EEG), with subsequent electromyography (EMG) on Day 10 showing reduced motor amplitudes (median nerve: right 0.64 mV, left 1.94 mV) and spontaneous fibrillations in limb muscles. Brain magnetic resonance imaging (MRI) demonstrated periventricular T1/T2 hyperintensities with diffusion restriction, though no structural anomalies were observed (Figure 2).
Axial MRI of the brain. The image demonstrates punctate lesions with short T1 and short T2 signal intensity (arrows) adjacent to the bilateral lateral ventricles. These lesions show restricted diffusion on diffusion weighted imaging (DWI) mapping.
Whole-exome sequencing (WES) was performed to investigate the underlying genetic etiology of the patient's severe neonatal presentation. WES identified a heterozygous missense variant in the ACTA1 gene, specifically at position c.227G>A, resulting in the amino acid substitution
p. Gly76Asp. This variant was not detected in either parent via Sanger sequencing, confirming its de novo origin. In accordance with the American College of Medical Genetics and Genomics (ACMG)/Association for Molecular Pathology (AMP) guidelines, the variant was classified as pathogenic. The evidence supporting this classification included the following: PS2 (de novo occurrence in a patient with the disease and no family history); PM1 (located in a mutational hot spot and/or critical functional domain well-established in disease pathology); PM2 (absent or very low frequency in population databases such as gnomAD); and PP3 (multiple computational predictions support a deleterious effect on the gene or gene product). Seen in Figure 3.
The validation results by sanger sequencing for the ACTA1 variants.
The infant demonstrated a complicated clinical course, requiring prolonged respiratory support—initially with 7 days of mechanical ventilation, followed by unsuccessful attempts at weaning to non-invasive positive pressure ventilation (NIPPV). Neuroprotective measures, including therapeutic hypothermia and vitamin B1/B12 supplementation, were implemented to mitigate neurological injury. Swallowing rehabilitation therapy was initiated but yielded minimal functional improvement. Although serial EEG monitoring showed gradual improvement by Day 7, the infant exhibited persistent, profound neuromuscular weakness inconsistent with typical hypoxic-ischemic encephalopathy recovery. Given the poor prognosis and lack of meaningful neurological recovery, the family elected to withdraw life-sustaining care on Day 18, and the infant passed shortly thereafter. A schematic overview of the clinical events development is provided in Figure 4.
The timeline for clinical events. CMV: conventional mechanical ventilation.
Discussion
This case illustrates a severe neonatal presentation of ACTA1-related myopathy, associated with a de novo heterozygous pathogenic variant (c.227G>A, p. Gly76Asp) in the ACTA1 gene. The infant exhibited classic features including profound hypotonia, respiratory failure, bulbar dysfunction, arthrogryposis, and an expressionless facies, consistent with previously reported cases of neonatal-onset actinopathy (7–11). Notably, the initial clinical picture complicated the diagnostic process due to significant overlap with hypoxic-ischemic encephalopathy (HIE), particularly given the observed perinatal asphyxia and metabolic acidosis. However, the persistence of severe neuromuscular weakness beyond the typical recovery window for HIE, along with the absence of significant hypoxic injury on neuroimaging, raised suspicion of an underlying neuromuscular disorder. This diagnostic challenge underscores the necessity of including genetic myopathies in the differential diagnosis of floppy infants, even with suggestive perinatal complications.
Genetic testing played a pivotal role in confirming the diagnosis. WES identified the missense variant p. Gly76Asp, affecting a highly conserved residue within the skeletal muscle α-actin protein. Previous functional studies of variants at this residue and adjacent amino acids have demonstrated detrimental effects on actin polymerization and filament stability, further corroborating its pathogenicity (12). The genetic inheritance of ACTA1-related disorders is complex, predominantly autosomal dominant. In this pattern, a pathogenic variant in a single allele (heterozygosity) is sufficient to result in the disease phenotype. This genetic finding is consistent with the severe clinical phenotype observed in the proband. In accordance with ACMG guidelines, the variant was classified as pathogenic based on several criteria: its de novo occurrence (PS2), low population frequency (PM2), localization within a critical functional domain (PM1), and supportive computational evidence of deleterious impact (PP3) (7, 13–17). The absence of the variant in both parents—confirmed via Sanger sequencing—further supports its de novo origin, which is a common inheritance pattern in severe neonatal cases (16, 17). The predominance of missense mutations, distributed throughout the gene without a clear hotspot, is well-documented in ACTA1-related disease, and this case reinforces that observation (7, 13–15).
The multisystemic manifestations observed here highlight the severe functional impact of ACTA1 mutations on striated muscle and related systems. Beyond generalized weakness and respiratory failure, the patient also showed elevated creatine kinase levels and cardiac ectopy, suggesting subclinical cardiac involvement—a finding occasionally noted in severe phenotypes (7, 9). Additionally, the combination of hypertonic contractures against a background of global hypotonia illustrates the complex and often paradoxical effects of actin dysfunction on muscle tone and contractility. Tragically, the clinical course in this case was fatal, consistent with the poor prognosis associated with neonatal-onset ACTA1 myopathies. As reported in the literature, such patients often experience rapid progression of respiratory insufficiency leading to early mortality, despite maximal respiratory and nutritional support (7, 11, 18). This outcome reinforces the devastating natural history of this condition and highlights the limited efficacy of current supportive interventions in altering disease progression in the most severe forms. The family's decision to withdraw life-sustaining care, though profoundly difficult, is not uncommon in such contexts and reflects the severe neurodevelopmental prognosis and lack of disease-modifying therapies (10, 11, 19).
This study has several limitations that warrant discussion. First, as a single-center case report, our conclusions are based on the observation of a solitary patient. Given the significant phenotypic heterogeneity associated with ACTA1-related myopathy, the generalizability of these findings to the broader patient population should be interpreted with caution. Second, this study lacks independent verification via muscle biopsy, which remains the traditional diagnostic gold standard. However, in the contemporary era of advanced molecular diagnostics, genetic testing has become a central pillar for the definitive diagnosis and classification of congenital myopathies, providing a reliable diagnostic pathway for critically ill patients where invasive biopsy is not feasible. Despite these limitations, the detailed clinical course and genetic information documented in this case provide a crucial natural history dataset and phenotypic framework. Current management of ACTA1-related myopathy remains primarily supportive; however, a deeper understanding of its pathological mechanisms—specifically delineating between loss-of-function and toxic gain-of-function effects—is driving a paradigm shift toward etiology-targeted treatments (1, 3). For loss-of-function mutations, adeno-associated virus (AAV)-mediated gene replacement therapy represents a highly promising strategy (20). The data from this report are essential for designing future clinical trials of such advanced therapies, particularly for application in the most severe neonatal population, as they can inform patient stratification, biomarker selection, and endpoint determination.
Conclusion
This case highlights the critical role of early genetic testing, such as rapid WES, in the diagnostic evaluation of neonates with unexplained hypotonia and respiratory failure. A definitive genetic diagnosis is essential to guide clinical management, facilitate accurate prognostic counseling, and inform family-centered decision-making. Future efforts should focus on elucidating genotype-phenotype correlations, developing targeted therapies, and establishing international registries to improve outcomes for patients with severe ACTA1-related congenital myopathies.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Zhang H Chang M Chen D Yang J Zhang Y Sun J Congenital myopathies: pathophysiological mechanisms and promising therapies. J Transl Med. (2024) 22(1):815. 10.1186/s 12967-024-05626-539223631 PMC 11370226 · doi ↗ · pubmed ↗
- 2Ogasawara M Nishino I. A review of major causative genes in congenital myopathies. J Hum Genet. (2023) 68(3):215–25. 10.1038/s 10038-022-01045-w 35668205 · doi ↗ · pubmed ↗
- 3Nowak KJ Ravenscroft G Laing NG. Skeletal muscle α-actin diseases (actinopathies): pathology and mechanisms. Acta Neuropathol. (2013) 125(1):19–32. 10.1007/s 00401-012-1019-z 22825594 · doi ↗ · pubmed ↗
- 4Zhang Y Yan H Liu J Yan H Ma Y Wei C Clinical and genetic features of infancy-onset congenital myopathies from a Chinese paediatric centre. BMC Pediatr. (2022) 22(1):65. 10.1186/s 12887-021-03024-035081925 PMC 8790871 · doi ↗ · pubmed ↗
- 5Ryan MM Schnell C Strickland CD Shield LK Morgan G Iannaccone ST Nemaline myopathy: a clinical study of 143 cases. Ann Neurol. (2001) 50(3):312–20. 10.1002/ana.108011558787 · doi ↗ · pubmed ↗
- 6Jungbluth H Treves S Zorzato F Sarkozy A Paolini C Orcesi S Congenital myopathies: disorders of excitation-contraction coupling and muscle contraction. Nat Rev Neurol. (2018) 14(3):151–67. 10.1038/nrneurol.2017.19129391587 · doi ↗ · pubmed ↗
- 7Laing NG Clarke NF Dye DE Liyanage K Walker KR Kobayashi Y Actin mutations are one cause of congenital fibre type disproportion. Ann Neurol. (2004) 56(5):689–94. 10.1002/ana.2026015468086 · doi ↗ · pubmed ↗
- 8Mulvany-Robbins B Putko B Schmitt L Oudit G Phan C Beecher G. Novel p.Asp 27Glu ACTA 1 variant features congenital myopathy with finger flexor weakness, cardiomyopathy, and cardiac conduction defects. Neuromuscul Disord. (2023) 33(7):546–50. 10.1016/j.nmd.2023.05.00937315422 · doi ↗ · pubmed ↗