Integrated fetal testicular transcriptomic and epigenomic profiles during maternal nutrient restriction with dietary melatonin intervention
Hala El Daous, Brittni P Littlejohn, Zully E Contreras-Correa, Shiveeli Rajput, Darcie R Sidelinger, E Heath King, Mark A Arick, Caleb O Lemley

TL;DR
This study shows how melatonin can help counteract the negative effects of poor maternal nutrition on fetal testis development by altering gene activity and DNA methylation.
Contribution
The study integrates transcriptomic and epigenomic data to reveal how melatonin supplementation affects fetal testis development under maternal nutrient restriction.
Findings
Melatonin supplementation altered gene expression and DNA methylation in fetal testes under nutrient restriction.
Specific genes like DAAM1 and KYAT1 showed significant expression changes with melatonin.
Nutrient restriction alone caused 370 DMRs without corresponding gene expression changes.
Abstract
Fetal development is a critical period that establishes reproductive efficiency and herd performance depending on in-utero epigenetic modifications. Dietary restrictions may affect fetal testis development and the offspring fertility. Several studies have connected genetic instability to circadian cycle disruptions, including epigenetic modifications to melatonin, a key regulator. On day 160 of gestation, 17 male-bearing Brangus heifers were assigned to one of four groups in a 2 × 2 factorial treatment arrangement: adequately fed (ADQ; 100% NRC recommendation, n = 3), nutrient restricted (RES; 60% NRC recommendation, n = 5), or ADQ or RES supplemented with 20 mg/d melatonin (ADQ-MEL, n = 5; RES-MEL, n = 4). On day 240 of gestation, heifers underwent Cesarean sections to collect fetuses and testicular tissues. The fetal testicular tissue was processed and analyzed using the…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
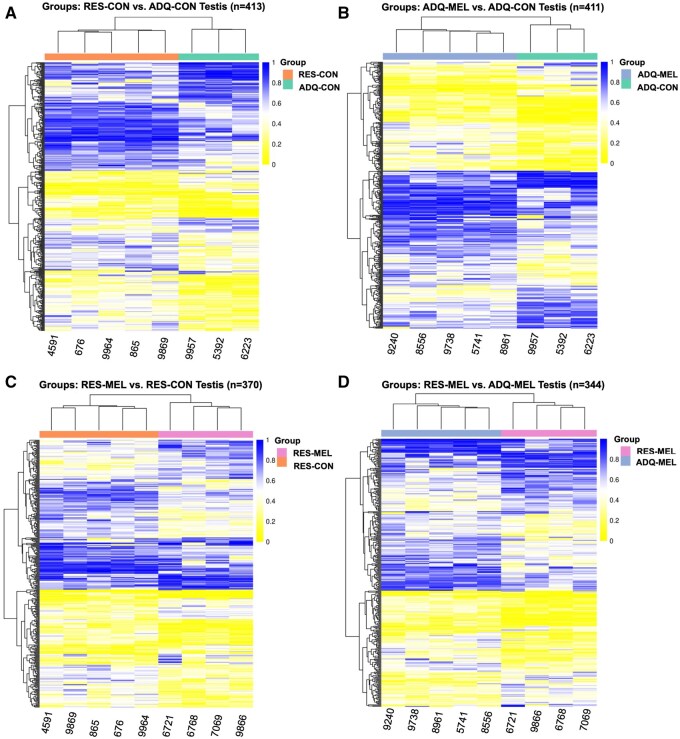 Figure 1
Figure 1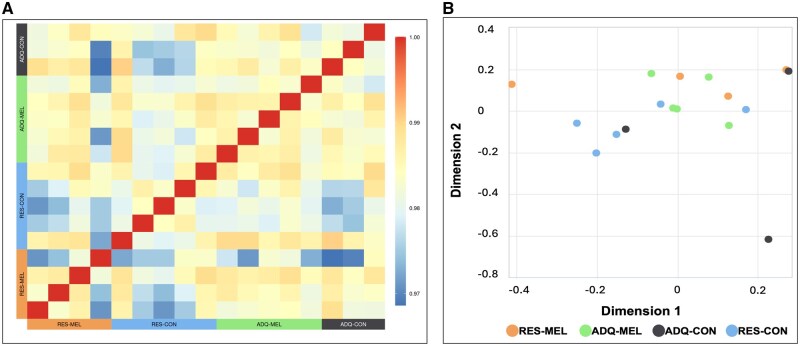 Figure 2
Figure 2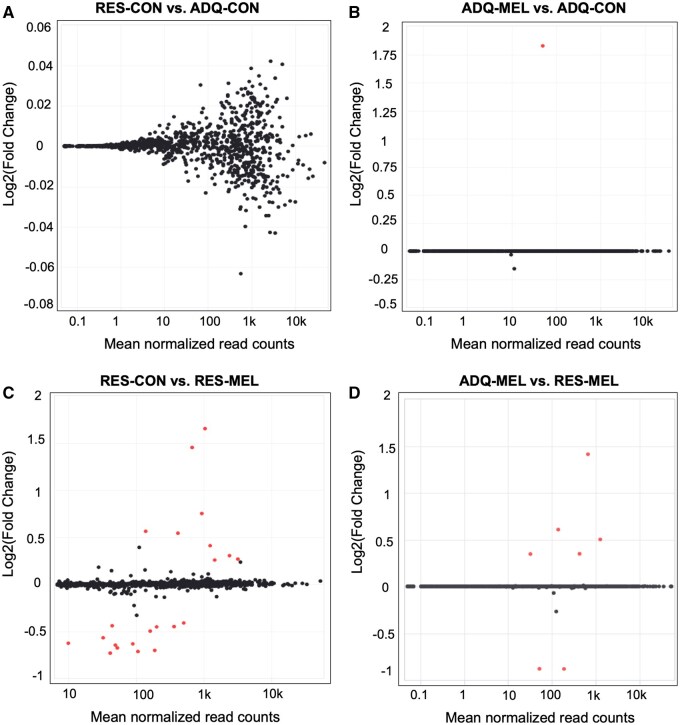 Figure 3
Figure 3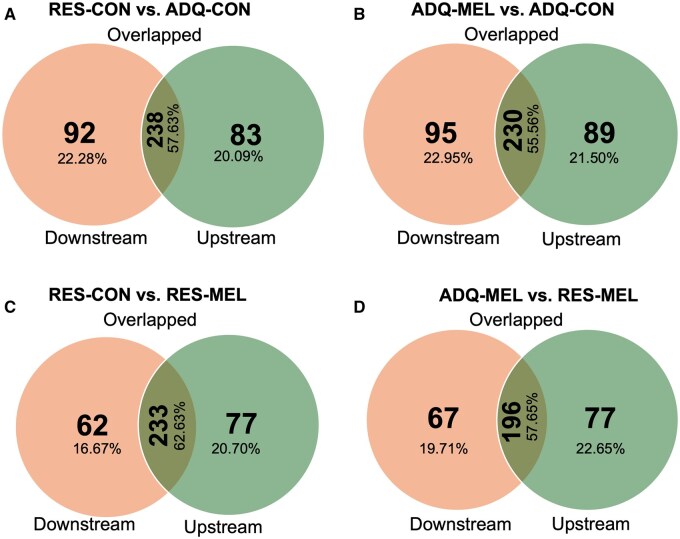 Figure 4
Figure 4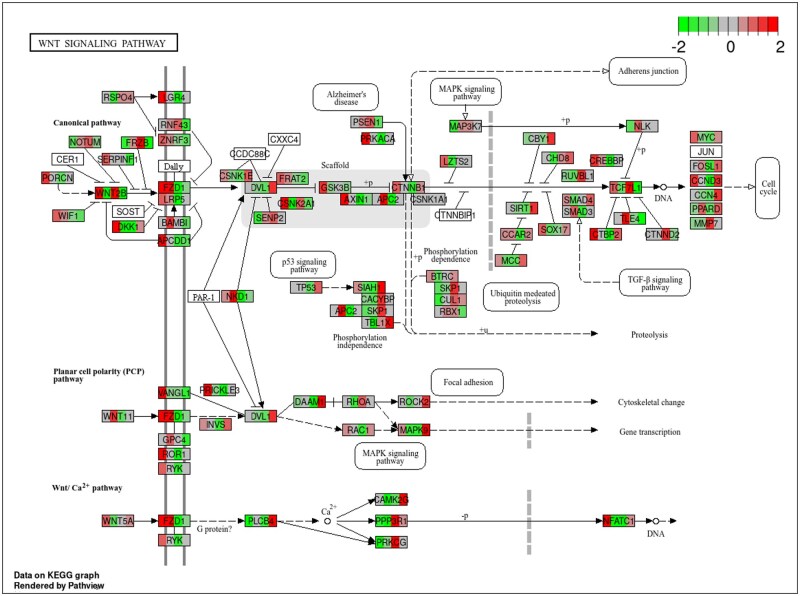 Figure 5
Figure 5| Type | Group | Input | Output |
|
|
|
|---|---|---|---|---|---|---|
|
| RES-MEL vs. RES-CON | 1,696,648 | 5,062 | 4,891 | 2,942 | 1,949 |
|
| RES-MEL vs. RES-CON | 1,696,648 | 370 | NA | 225 | 145 |
|
| ADQ-MEL vs. ADQ-CON | 1,571,549 | 5,824 | 5,558 | 2,010 | 3,548 |
|
| ADQ-MEL vs. ADQ-CON | 1,571,549 | 411 | NA | 142 | 269 |
|
| RES-CON vs. ADQ-CON | 1,600,570 | 5,714 | 5,418 | 1,756 | 3,662 |
|
| RES-CON vs. ADQ-CON | 1,600,570 | 413 | NA | 127 | 285 |
|
| RES-MEL vs. ADQ-MEL | 1,666,164 | 4,590 | 4,407 | 2,656 | 1,751 |
|
| RES-MEL vs. ADQ-MEL | 1,666,164 | 344 | NA | 210 | 133 |
| Comparisons | Promoters | Exons | Introns | GpC islands |
|---|---|---|---|---|
|
| 25 | 89 | 183 | 83 |
|
| 18 | 94 | 219 | 117 |
|
| 32 | 97 | 219 | 114 |
|
| 24 | 99 | 189 | 116 |
| Treatment | GO: term name | Query | Term size | GO: Term ID | Source |
|---|---|---|---|---|---|
|
| Regulation of signaling | 1.57E−06 | 2591 | GO: 0023051 | GO: Biological Process |
| Regulation of cell communication | 4.03E−06 | 2587 | GO: 0010646 | GO: Biological Process | |
| Biological regulation | 9.69E−05 | 10961 | GO: 0065007 | GO: Biological Process | |
| Anatomical structure development | 1.04E−04 | 4390 | GO: 0048856 | GO: Biological Process | |
| Multicellular organism development | 1.67E−04 | 3409 | GO: 0007275 | GO: Biological Process | |
| Developmental process | 3.31E−04 | 4730 | GO: 0032502 | GO: Biological Process | |
| Cell–cell signaling | 3.40E−04 | 1149 | GO: 0007267 | GO: Biological Process | |
| Cell adhesion | 6.90E−04 | 1073 | GO: 0007155 | GO: Biological Process | |
| Regulation of biological process | 8.22E−04 | 10636 | GO: 0050789 | GO: Biological Process | |
| Regulation of signal transduction | 1.01E−03 | 2252 | GO: 0009966 | GO: Biological Process | |
| Positive regulation of biological process | 1.60E−03 | 4972 | GO: 0048518 | GO: Biological Process | |
| Regulation of cellular process | 1.70E−03 | 10155 | GO: 0050794 | GO: Biological Process | |
| Positive regulation of cellular process | 3.96E−03 | 4528 | GO: 0048522 | GO: Biological Process | |
| Signaling | 4.69E−03 | 5732 | GO: 0023052 | GO: Biological Process | |
| Cell communication | 4.73E−03 | 5814 | GO: 0007154 | GO: Biological Process | |
| Cell junction organization | 8.49E−03 | 556 | GO: 0034330 | GO: Biological Process | |
| Cell differentiation | 1.03E−02 | 3138 | GO: 0030154 | GO: Biological Process | |
| Cellular developmental process | 1.03E−02 | 3138 | GO: 0048869 | GO: Biological Process | |
| RHOA GTPase cycle | 1.68E−02 | 128 | REAC: R-BTA-8980692 | Reactome | |
| Regulation of response to stimulus | 2.46E−02 | 3007 | GO: 0048583 | GO: Biological Process | |
| System development | 2.53E−02 | 2867 | GO: 0048731 | GO: Biological Process | |
| Cell junction assembly | 2.96E−02 | 321 | GO: 0034329 | GO: Biological Process | |
| Nervous system development | 3.14E−02 | 1727 | GO: 0007399 | GO: Biological Process | |
| Negative regulation of signal transduction | 3.41E−02 | 995 | GO: 0009968 | GO: Biological Process | |
| Regulation of cell morphogenesis | 4.91E−02 | 174 | GO: 0022604 | GO: Biological Process | |
|
| Positive regulation of biological process | 5.76E−03 | 4972 | GO: 0048518 | GO: Biological Process |
| Regulation of biological process | 6.02E−03 | 10636 | GO: 0050789 | GO: Biological Process | |
| Biological regulation | 1.04E−02 | 10961 | GO: 0065007 | GO: Biological Process | |
| Regulation of developmental process | 1.19E−02 | 1826 | GO: 0050793 | GO: Biological Process | |
| Regulation of cell communication | 1.78E−02 | 2587 | GO: 0010646 | GO: Biological Process | |
| Regulation of signaling | 1.85E−02 | 2591 | GO: 0023051 | GO: Biological Process | |
| Regulation of response to stimulus | 3.75E−02 | 3007 | GO: 0048583 | GO: Biological Process | |
| Endocytosis | 4.82E−02 | 230 | KEGG: 04144 | KEGG Pathway | |
|
| Animal organ development | 3.13E−03 | 2214 | GO: 0048513 | GO: Biological Process |
| Positive regulation of biological process | 1.22E−02 | 4972 | GO: 0048518 | GO: Biological Process | |
|
| Phosphorus metabolic process | 2.72E−02 | 1930 | GO: 0006793 | GO: Biological Process |
| Regulation of signaling | 2.92E−02 | 2591 | GO: 0023051 | GO: Biological Process | |
| Phosphate-containing compound metabolic process | 4.75E−02 | 1906 | GO: 0006796 | GO: Biological Process |
| Comparison | Gene ID | Gene abbreviation | log2FoldChange | Adjusted |
|---|---|---|---|---|
|
| ENSBTAG00000024420 | COL28A1 | 1.9216 | 9.41E−07 |
| ENSBTAG00000022275 | RPL10 | 1.7814 | 5.01E−05 | |
| ENSBTAG00000050253 | TRPM3 | 0.5793 | 0.0002 | |
| ENSBTAG00000012848 | PTPRU | −0.8186 | 0.0023 | |
| ENSBTAG00000038168 | SLITRK1 | 0.8908 | 0.0023 | |
| ENSBTAG00000013401 | ARHGEF40 | 0.3287 | 0.0053 | |
| ENSBTAG00000034693 | SYT1 | 0.4500 | 0.0122 | |
| ENSBTAG00000049347 | LOC112442676 | −4.0405 | 0.0170 | |
| ENSBTAG00000007698 | TMEM59L | −0.9603 | 0.0196 | |
| ENSBTAG00000005984 | DAAM1 | 0.3202 | 0.0267 | |
| ENSBTAG00000008274 | ENSBTAG00000008274 | −1.5941 | 0.0285 | |
| ENSBTAG00000024086 | TMEM35B | 0.7129 | 0.0285 | |
| ENSBTAG00000002999 | ANAPC15 | −0.5421 | 0.0285 | |
| ENSBTAG00000003200 | ENSBTAG00000003200 | 0.3188 | 0.0285 | |
| ENSBTAG00000018064 | FAM221A | −1.0836 | 0.0329 | |
| ENSBTAG00000031238 | SHCBP1L | −0.9366 | 0.0338 | |
| ENSBTAG00000020352 | PAQR5 | −1.0338 | 0.0354 | |
| ENSBTAG00000008511 | PPP4R3C | −1.7128 | 0.0407 | |
| ENSBTAG00000021072 | DTNB | −0.5280 | 0.0450 | |
| ENSBTAG00000051764 | LncRNA | −1.9899 | 0.0463 | |
| ENSBTAG00000003171 | SHANK2 | −0.7015 | 0.0489 | |
| ENSBTAG00000004863 | RIC3 | −0.6131 | 0.0489 | |
|
| ENSBTAG00000012848 | PTPRU | −0.9522 | 0.0001 |
| ENSBTAG00000022275 | RPL10 | 1.7182 | 0.0002 | |
| ENSBTAG00000034693 | SYT1 | 0.5203 | 0.0011 | |
| ENSBTAG00000011881 | TDRD10 | −1.3270 | 0.0290 | |
| ENSBTAG00000024086 | TMEM35B | 0.7599 | 0.0338 | |
| ENSBTAG00000052713 | H2B | 2.9672 | 0.0493 | |
| ENSBTAG00000050253 | TRPM3 | 0.4598 | 0.0493 |
- —National Institute of Food and Agriculture10.13039/100005825
- —U.S. Department of Agriculture10.13039/100000199
- —U.S. Department of Agriculture, Agricultural Research Service10.13039/100007917
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsReproductive Physiology in Livestock · Birth, Development, and Health · Circadian rhythm and melatonin
Introduction
The fetal programming hypothesis suggests that stimuli that occurs during a sensitive period of development can program fetal tissues and exert organizational effects that continue throughout the offspring’s life (Vonnahme and Lemley, 2011; Lemley, 2020). It is increasingly apparent that environmental alterations, including nutritional modifications, can lead to variations in offspring growth, metabolism, reproduction, and health in later life (Caton et al., 2019). The nutritional status of the dam is crucial throughout pregnancy, as external stimuli or perturbations during gestation may result in phenotypic and metabolic alterations in the growing fetus (Reynolds et al., 2019). Beef cattle, when reared on pasture, are particularly vulnerable to nutritional deficiencies caused by seasonal variations in forage quality. Numerous studies have demonstrated the impact of maternal diet on the development of muscle and adipose tissue (Costa et al., 2021; Polizel et al., 2021; Zhang et al., 2021), and body weight (Long et al., 2021); however, research concerning the influence of maternal nutrition on the performance, metabolism, and health of male replacement animals is scarce. Nutrient restriction throughout testis development in utero may affect testis function and subsequently contribute to subfertility in mature males (McCoski et al., 2021).
Epigenetic modifications that result from alternations in prenatal nutrition during crucial developmental periods may permanently alter physiology and metabolism (Godfrey and Barker, 2001; Yan et al., 2013). Maternal nutrition affects fetal development through modifying epigenetic markers that regulate gene expression without changing DNA sequence (Lan et al., 2013). These epigenetic markers include DNA methylation, histone changes, and non-coding RNA activity (Jaenisch and Bird, 2003; Ji et al., 2016). DNA methylation regulates transcription, genomic imprinting, and X chromosome inactivation in mammals (Suzuki and Bird, 2008; Pelizzola and Ecker, 2011). Changes in maternal nutrient availability can affect fetal genome methylation by altering DNA methylation enzymes or by modifying substrate availability (Choi and Friso, 2010; Ji et al., 2016).
Melatonin has recently emerged as a potential therapeutic in alleviating adverse environmental effects such as maternal undernutrition in both sheep and cattle (Lemley et al., 2012, 2013; Contreras-Correa et al., 2021, 2022). Melatonin is known as a powerful antioxidant and blood flow regulator that can pass through the placenta into ovine and bovine fetal circulation (Lemley et al., 2012; Brockus et al., 2016). Interestingly, Contreras-Correa et al. (2021) reported that nutrient restriction decreased uterine blood flow (UBF) in pregnant Brangus heifers and melatonin supplementation increased UBF depending on the season. Melatonin has also been shown to reduce severe DNA damage, a consequence of unstable oxygen-based reactants in rats and mice models (Reiter et al., 2004) and can serve as an epigenetic modifier in human (Korkmaz and Reiter, 2008). In addition to the positive effects melatonin has on female reproductive performance, melatonin may also be a beneficial modulator of male reproductive efficiency. McCarty et al. (2018) reported increased scrotal circumference and testicular weight at the time of weaning in bull calves born from melatonin supplemented beef cattle (two subdermal ear implants containing 24 mg of melatonin) at day 180, 210, and 240 of gestation. Melatonin receptors have been found in Sertoli and Leydig cells in the testes (Frungieri et al., 2017; González-Arto et al., 2017), and exogenous melatonin upregulates gene expression of spermatogenesis-related genes (Yang et al., 2014). Despite evidence that melatonin may affect male offspring performance, the combined effects of melatonin supplementation and nutrient restriction on testicular DNA methylation and gene expression related to testis development, spermatogenesis, and fertility are still poorly understood.
We hypothesized that melatonin supplementation and a nutritional deficit throughout mid-to-late gestation would affect fetal testis DNA methylation and thereby transcriptomic profiles. We aimed to determine the differentially methylated regions (DMRs) in fetal testis tissue from nutritionally restricted or adequately fed dams that had been supplemented with melatonin or a vehicle control. Additionally, the association between transcriptome patterns and DNA methylation signatures was examined to investigate possible outcomes related to bull reproductive efficiency.
Materials and Methods
Animal procedure and tissue collection
The Institutional Animal Care and Use Committee at Mississippi State University approved the use and care of the study animals (#17-709). All animal breeding, diets, treatments, and Caesarean sections procedures were previously described by Contreras-Correa et al. (2021). In summary, this study included only male-bearing, spring calving Brangus heifers (n = 17) [ADQ-CON (n = 3), RES-CON (n = 5), ADQ-MEL (n = 5), and RES-MEL (n = 4)], that were bred to a single sire by artificial insemination. The heifers were anticipated to give birth in January 2020. In a 2 × 2 factorial design, heifers were stratified by body weight at day 160 of gestation and randomly assigned to one of four dietary treatments: adequately fed (ADQ-CON; 100% NRC recommendation), nutrient restricted (RES-CON, 60% NRC recommendation), or adequately fed or nutrient restricted supplemented with 20 mg/d of melatonin (ADQ-MEL; RES-MEL), or adequately fed (ADQ-CON; 100% NRC recommendation) (NRC, 2000). Melatonin (#14427; Cayman Chemical Company, Ann Arbor, MI, USA) feeding and dietary composition have already been discussed (Contreras-Correa et al., 2021), animals were supplemented with 20 mg/d of melatonin dissolved in 2 mL of absolute ethanol, while plain absolute ethanol served as vehicle control (CON). The heifers were fed a grain mix top dressed in the treatment CON or MEL at 0900 h, and after consuming their treatments, they were given a total mixed ration ADQ or RES using the Calan gates electronic feeding system. Weekly dietary adjustments were made in accordance with the dam’s body weight.
Cesarean section and fetal tissue collection
According to Contreras-Correa et al. (2021) heifers were subjected to Cesarean procedures on day 240 of gestation to collect fetal tissue. A Silencer hydraulic squeeze chute was used to restrain the animals during surgeries at the H. H. Leveck Animal Research Center. Aseptic surgery was performed on the skin surrounding the incision site after a paravertebral or inverted-L block with 2% lidocaine. An incision of 20 cm was performed ventral to the paralumbar fossa’s transverse processes. The gravid uterus was delivered to the incision site using the fetus’s frontal limbs, which were identified and employed as a handle. The umbilical cord was identified, clamped, and cut following a uterine incision. For 4 wk following surgery, a veterinarian checked the cattle every day for clinical indications of infection. Following fetal extraction, testicular parenchymal tissue was harvested, deposited in cryogenic tubes, rapidly frozen in liquid nitrogen, and preserved at −80°C for subsequent analysis. Fetal testicular parenchymal tissue was sent to Zymo Research Corp (Zymo Research, Irvine, CA) for the investigation of DNA methylation and RNA transcriptome.
Methyl-MiniSeq Genome-Wide bisulfite sequencing (GWBS)
Genomic DNA was isolated from the 17 fetal testicular specimens with the Quick-DNA/RNA Miniprep Plus Kit (Cat#: D7003, Zymo Research). The DNA samples were used for processing and analysis. Briefly, the Zymo Research DNA Clean & Concentrator™-5 (Cat#: D4003) was used to purify 500 ng of input genomic DNA after it had been successively digested with 60 units of Taqαl and 30 units of MspI (NEB). Following Illumina’s instructions, fragments were ligated to pre-annealed adapters that included 5ʹ-methyl-cytosine rather than cytosine. Using a 2.5% NuSieve 1:1 agarose gel, adaptor-ligated fragments ranging in size from 150–250 bp and 250–350 bp were extracted using the Zymoclean^TM^ Gel DNA Recovery Kit (Cat#: D4001). After that, the EZ DNA Methylation-Lightning^TM^ Kit (Cat#: D5030) was used to bisulfite-treat the fragments, and DNA Clean & Concentrator™-5 (Cat#: D4003) was used to purify the products of preparative-scale PCR. Methyl-MiniSeq libraries were sequenced using the Illumina, NovaSeq 6000 (San Diego, CA). TrimGalore 0.6.4 was then used to modify raw FASTQ files, fill in nucleotides, and trim quality. The impact of trimming and the overall quality distributions of the data were evaluated using FastQC 0.11.9. Trimmed reads were aligned to the Bos taurus ARS-UCD v1.2 reference genome (GCF_002263795.1) using Bismark 0.22.3. MethylDackel 0.5.0 was used to call the methylation and unmethylated read totals for every CpG site. Differentially methylated cytosines (DMCs) and regions (DMRs) were identified, annotated, and visualized using DSS through a comparative statistical analysis. Cytosines with read depth ≥ 5 in ≥ 2 samples per group were retained. DSS, which by default performed the Wald test and the Benjamini-Hochberg P-value adjustment, was used to identify DMCs and DMRs. FDR ≤ 0.05 and absolute methylation difference > 0.1 are considered significant DMCs and DMRs. Genes overlapping the DMRs were then subjected to a functional enrichment analysis using the g: Profiler tool. The DMCs and DMRs were also annotated with the closest expressed gene in the transcriptome dataset using bedtools v2.31.1 (https://doi.org/10.1093/bioinformatics/btq033) (Quinlan and Hall, 2010).
Whole transcriptome analysis of fetal testicular tissue
The quick-RNA Mininprep Plus Kit (Cat#: R1057; Zymo Research) was used for isolation and purification of total RNA from the 17 fetal testicular tissue specimens. Libraries were constructed utilizing the Zymo-Seq RiboFree Total RNA Library Prep Kit (Cat # R3000) in accordance with the manufacturer’s instruction (v1.3.0). Briefly, RNA underwent reverse transcription to form cDNA, subsequently followed by the depletion of ribosomal RNA. Subsequently, the partial P7 adapter sequence was ligated to the 3ʹ end of cDNAs, followed by second strand synthesis and ligation of the partial P5 adapter to the 5ʹ end of the double-stranded DNAs. Finally, libraries were enhanced to include full-length adapters. The successful construction of the library was validated using Agilent’s D1000 ScreenTape Assay on the TapeStation. RNA-Seq libraries were sequenced on an Illumina platform to a minimum depth of 30 million read pairs per sample. The RNA-seq data were analyzed using the Zymo Research RNA-Seq pipeline, originally adapted from nf-core/rnaseq pipeline v1.4.2 (https://github.com/nf-core/rnaseq) (Patel et al., 2020) using Nextflow (https://www.nextflow.io/) (Di Tommaso et al., 2017). Briefly, quality control of raw reads was carried out using FastQC v0.11.9 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc). Adapter and low-quality sequences were trimmed from raw reads using Trim Galore! v0.6.6 (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore). Trimmed reads were aligned to the Bos taurus ARS-UCD v1.2 reference genome (Ensemble release 110, https://doi.org/10.1093/nar/gkad1049) (Peter et al., 2024) using STAR v2.6.1d (https://github.com/alexdobin/STAR) (Dobin et al., 2013). BAM file fconceptiltering and indexing was carried out using SAMtools v1.9 (https://github.com/samtools/samtools) (Danecek et al., 2021). RNAseq library quality control was implemented using RSeQC v4.0.0 (http://rseqc.sourceforge.net/) (Wang et al., 2012) and QualiMap v2.2.2-dev (http://qualimap.conesalab.org/) (García-Alcalde et al., 2012). Duplicate reads were marked using Picard tools v2.23.9 (http://broadinstitute.github.io/picard/) (Picard Toolkit, 2019). Library complexity was estimated using Preseq v2.0.3 (https://github.com/smithlabcode/preseq) (Daley and Smith, 2013). Duplication rate quality control was performed using dupRadar v1.18.0 (https://bioconductor.org/packages/dupRadar/) (Sayols et al., 2016). Reads overlapping with exons were assigned to genes using featureCounts v2.0.1 (http://bioinf.wehi.edu.au/featureCounts/) (Liao et al., 2014). Differential gene expression analysis was completed using DESeq2 v1.28.0 (https://bioconductor.org/packages/DESeq2/) (Love et al., 2014). Functional enrichment analysis was achieved using g: Profiler python API v1.0.0 (https://biit.cs.ut.ee/gprofiler/gost) (Raudvere et al., 2019). Quality control and analysis results plots were visualized using MultiQC v1.9 (https://github.com/ewels/MultiQC) (Ewels et al., 2016).
Overlapping analysis of DNA methylation and gene expression
We investigated the relationship between DNA methylation and differential gene expression to evaluate the genomic effects of maternal dietary restriction, both with and without melatonin administration. The common genes between DMRs and DEGs across different treatment groups were identified by an overlap analysis using bedtools v2.31.1 (https://doi.org/10.1093/bioinformatics/btq033). The diagrams were made by the Venny 2.1 Online tool (Venny, 2007).
Results
Methyl-MiniSeq Genome-wide bisulfite sequencing (GWBS) of fetal testicular tissue
Sequencing of the bisulfite libraries from 17 fetal testicular tissue samples, resulted in an average CpG coverage of 13.26 million reads (ranging from 9.11 to 19.284 million reads). After trimming and mapping, 2,768,233 total methylated sites were found. The distribution of methylation values for all samples is illustrated in Supplementary Figure S1A, and the distribution of mean methylation values for each treatment group are illustrated in Supplementary Figure S1B. The four contrasts (RES-MEL vs. RES-CON, ADQ-MEL vs. ADQ-CON, RES-CON vs. ADQ-CON, and RES-MEL vs. ADQ-MEL) were used to examine DMCs, resulting in 4,891, 5,558, 5,418, and 4,407 significant DMCs (FDR < 0.05), respectively (Table 1). DMCs that are near each other are grouped into differentially methylated regions (DMRs), resulting in 413 significant DMRs for RES-CON vs. ADQ-CON, the highest DMRs number in between the treated groups (Figure 1A); 411 for ADQ-MEL vs. ADQ-CON (Figure 1B); 370 significant DMRs for RES-MEL vs. RES-CON (Figure 1C); and 344 for RES-MEL vs. ADQ-MEL (Figure 1D).
Heatmap showing the hierarchical clustering of differential methylation in the genomic regions of fetal testicular tissue among different treatment groups. (A) Differentially methylated regions (DMRs; n = 413) between RES-CON (orange) and ADQ-CON (green), (B) ADQ-MEL (grey) and ADQ-CON (green; n = 411), (C) RES-CON (orange) and RES-MEL (pink; n = 370), and (D) ADQ-MEL (gray) and RES-MEL (pink; n = 344). Significant DMRs have FDR ≤ 0.05 and absolute methylation difference ≥ 0.1. The row represents the gene, and the column denotes the sample. Yellow: hypomethylated genes; blue: hypermethylated genes. The darkness of each color corresponds to the magnitude of the differences vs. the mean value.
To further evaluate the genomic effects of nutritional restriction with and without melatonin supplementation on fetal testis development, we investigated the relationship between DNA methylation, gene annotations, and gene expression profiles. Differentially methylated genes (DMGs) were found by overlapping DMRs with annotated genomic regions, including genes, exons, introns, promoters, and CpG islands. The majority of the DMRs are located within the introns followed by CpG islands, exons, and promoters of annotated genes as shown in Table 2.
Functional enrichment of DMGs
DMRs in the promoter region have the capacity to affect gene transcription, whereas DMRs in the gene body region frequently exhibit a positive correlation with gene expression levels. This analysis of these DMGs can offer valuable insights into the regulatory functions of DNA methylation. Comparing RES-CON vs. ADQ-CON testicular tissues, there were 417 DMGs (Supplementary Table S1). Many of the top significant Gene Ontology (GO) terms appear to be involved in fetal testicular development as a main effect of nutrient restriction during mid and late gestation (examples included: anatomical structure development, multicellular organism development, developmental process, cell differentiation, system development and regulation of cell morphogenesis alongside with catalytic activities, as shown in Table 3). When comparing ADQ-MEL with ADQ-CON 420 DMGs were identified, some of which were hypomethylated (such as: Scavenger receptor class F member 1 [SCARF1], non-specific serine/threonine protein kinase [TRIO], and RNA binding protein fox-1 homolog 3 [RBFOX3] and others were hypermethylated (such as: Dachsous cadherin-related 2 [DCHS2], Collectin-11 [COLEC11], Tubulin tyrosine ligase like 8 [TTLL8], and Palladin [PALLD]) (Supplementary Table S2). These previously listed genes were involved mainly in animal organ development, positive regulation of biological process as reproductive process. These results indicated that nutritional alongside with melatonin supplementation are important for animal organs development. The contrast RES-MEL vs. RES-CON contained 377 DMGs. These DMGs were significantly enriched in GO terms such as phosphorus metabolic process, developmental process, and response to stimulus (Supplementary Table S3). Comparing RES-MEL with ADQ-MEL results in 348 DMGs, significantly enriched in positive regulation of biological process, regulation of developmental process, and regulation of response to stimulus (Table 3). In comparison between RES-MEL and ADQ-MEL, out of 345 differentially methylated genes, 196 exhibited overlaps (Supplementary Table S4). The majority of these genes participate in the regulation of biological and developmental processes, as well as responses to stimuli as designed in our study for maternal nutrition restriction during mid-late gestation (Table 3).
Whole transcriptome analysis of fetal testicular tissue
Transcriptome analysis was performed on 17 fetal testicular samples. The RNA-seq libraries were sequenced to an average depth of 46.17 million reads per sample (ranging from 38M in S5741 to 55.5M in S8556) with 48% average GC%. Trimming removed and average of 0.23% reads per sample. An average 99.77% (ranging from 99.70% to 99.80%) of the trimmed data mapped to the reference genome. After QC of these alignments, feature counts found 25,706 genes expressed. Correlations between each fetal testicular tissue sample are visualized in a heatmap (Figure 2A). Additionally, multidimensional scaling was conducted to visualize the distance/similarity between samples using the 500 genes with highest variance (Figure 2B).
(A) Similarity matrix of fetal testicular tissue samples, the similarities were calculated using normalized and ‘rlog’ transformed read counts of all genes using DESeq2, minimum was 0.97 and maximum was 1.0. (B) Multidimensional scaling analysis of fetal testicular samples, adequately fed (ADQ-CON; 100% NRC recommendation, S9957, S6223, S5392), nutrient restricted (RES-CON, 60% NRC recommendation, S4591, S9964, S676, S9869, S865), and adequately fed or nutrient restricted supplemented with 20 mg/d of melatonin (ADQ-MEL, S8556, S5741, S9240, S9738, S8961; MEL-RES, S6721, S6768, S7059, S9866).
DESeq2 was used to conduct statistical analysis of differential gene expression. Genes with Benjamini-Hochberg adjusted P-values (FDR) < 0.05 were considered differentially expressed. When comparing RES-CON with ADQ-CON (Supplementary Table S5), no significantly differentially expressed genes were found (Figure 3A). The ADQ-MEL vs. ADQ-CON comparison (Supplementary Table S6) had a single significantly (FDR = 0.0005) upregulated (log2 FC = 1.864) gene: Kynurenine—oxoglutarate transaminase 1 (KYAT1) (ENSBTAG00000036099; Figure 3B). In contrast, comparing RES-MEL and RES-CON (Supplementary Table S7) reveals 9 upregulated and 13 downregulated genes (Table 4; Figure 3C). Finally, five genes were upregulated and two downregulated in RES-MEL versus ADQ-MEL (Table 4; Figure 3D, Supplementary Table S8).
The MA plots for differentially expressed genes (up to the first 1,000 genes) and up to 1,000 randomly selected non-differentially expressed genes. Expression levels are shown on X-axis while Log2 of fold changes (Log2 FC) are shown on Y-axis. Red dots represent differentially expressed genes (adjusted P-values < 0.05) comparing between RES-CON and ADQ-CON testicular tissues (A), ADQ-MEL vs. ADQ-CON testicular tissue (B), RES-CON vs. RES-MEL (C), ADQ-MEL vs. RES-MEL (D). Grey dots represent non-differentially expressed genes.
Overlapping analysis of DNA methylation and gene expression
To assess the genomic impacts of maternal nutritional restriction, both with and without melatonin supplementation, we examined the correlation between DNA methylation and gene expression. An overlap analysis was performed to determine the common genes between DMRs and DEGs across various treatment groups. We identified 238 (57.63%) overlapping genes between the RES-CON vs. ADQ-CON fetal testicular tissue, 230 (55.56%) overlapping genes in the ADQ-MEL vs. ADQ-CON fetal testicular tissue, the highest percentage (62.63%) of overlapped genes (233) were identified for RES-CON vs. RES-MEL fetal testicular tissue, and 196 genes in the ADQ-MEL vs. RES-MEL fetal testicular tissue (Figure 4A–D). The detailed list for overlapped genes for all treated groups is listed in Supplementary Tables S1–S4. Interestingly, Disheveled-associated activator of morphogenesis 1 (DAAM1), in RES-MEL vs. RES-CON, was the only overlapped statistically significant differentially expressed gene DEG (log2 FC = 0.320) that was also differentially methylated (methylation difference −0.2091) (Supplementary Table S3). Interestingly, this gene is involved in Wnt pathway as illustrated in Figure 5.
Venn diagrams for the numbers and locations of methylated regions of the expressed genes. The orange color indicated that the methylation region is downstream of the gene, while the green color indicated that the methylation region is upstream of the gene, the olive green means the expressed gene and methylation region overlap for each comparsion. (A) RES-CON vs. ADQ-CON; (B) ADQ-MEL vs. ADQ-CON; (C) RES-CON vs. RES-MEL; (D) ADQ-MEL vs. RES-MEL.
WNT signaling pathway with the expression data mapped to it. Pathway information generated by Kyoto Encyclopedia of Genes and Genomes (KEGG). The graph shows the gene-wise normalized expression (z score) for each group (ADQ-CON, ADQ-MEL, RES-CON, and RES-MEL). Because of the normalization, the expression data is comparable within a gene but not across genes.
Discussion
Although it is well known that maternal nutrition can affect fetal development in both males and females, most recent studies focus mainly on female offsprings. Consequently, male developmental programming remains inadequately comprehended. Numerous maladaptive characteristics may result from inadequate maternal nutrition in offspring. Therefore, this investigation postulated that the epigenomic and transcriptome patterns of fetal testicular tissue, and the developmental programming of male fetuses, would be altered by maternal nutrient restriction and melatonin supplementation.
In ruminants 60% to 90% of fetal growth occurs during the last third of gestation (Reynolds and Redmer, 1995). Male offspring exhibit elevated energy requirements for growth and may be more susceptible to maternal nutrition restriction (Hewison and Gaillard, 1999). Bull Sertoli cell counts rise from mid-gestation until birth (Hochereau-de Reviers et al., 1987) and continue to rise through puberty (Andrade et al., 2013), rendering maternal nutritional restriction during this window more detrimental than early gestational intervention. In this study we commenced our experiment around mid to late gestation, specifically on day 160, to align with testicular function and sexual differentiation, ending on day 240 of gestation. Melatonin diminishes testicular reactive oxygen species (ROS) and inflammation (Frungieri et al., 2017, El Shalofy et al., 2021). Melatonin upregulated spermatogenesis-related genes like Cyclin D1, Cyclin E, Dhh, Occludin, and Claudin in bovine Sertoli cells (Yang et al., 2014). Melatonin suppresses 8-OH-deoxyguanosine synthesis, activates p53, and enhances the expression of repair and anti-apoptotic proteins, thereby protecting DNA (Slominski et al., 2017; Skobowiat, 2018). Melatonin has the potential to boost the embryonic development progress through several mechanisms including DNA demethylation of pluripotency related gene (Oct4), DNA methylation maintenance of imprinted gene H19/Igf2, and the remethylation of tissue-specific gene Thy1 in cloned embryos (Qu et al., 2020). It is well known that DNA hypermethylation in the promoter regions silencing gene expression. However, Namous et al. (2018) also found that DNA hypermethylation in the gene body influences expression either by silencing or enhancement. In this study most of the DMRs were found in the introns, then the exons, and lastly the promoters of DMGs. These distinct methylation alteration sites identified in this study influenced the significant expression patterns via either upregulation or downregulation. Comparing RES-CON with ADQ-CON, exhibited the highest DMRs frequency among the treated groups. It has been shown that a dam’s diet can alter the epigenetic status of her offspring during embryonic development.
An integrative analysis of the methylome and transcriptome profiles of fetal testicular tissues revealed a remarkable overlap between DMGs and DEGs. Interestingly, a comparison between RES-MEL and RES-CON indicated the key roles of melatonin supplementation in nutritionally restricted dams and compromised pregnancies. Nine genes (DAAM1, Collagen alpha-1(XXVIII) chain (COL28A1), Large ribosomal subunit protein uL16 (RPL10), Transient receptor potential cation channel subfamily M member 3 (TRPM3), SLIT and NTRK like family member 1 (SLITRK1), Rho guanine nucleotide exchange factor 40 (ARHGEF40), Synaptotagmin-1 (SYT1), Transmembrane protein 35B (TMEM35B), and chondroitin sulfate proteoglycan family member 4B (CSPG4B) demonstrate significantly elevated expression in the RES-MEL. Among these significant genes, only the DAAM1 gene, located on chromosome 10, was hypomethylated. Other genes overlapped by DMRs include the hypomethylated genes Diacylglycerol kinase (DGKD), H2.0-like homeobox protein (HLX), Follistatin like 4 (FSTL4), histone H3-lysine (9) N-methyltransferase (EHMT)1, and TNF receptor superfamily member 19 (TNFRSF19), and hypermethylated genes Palmitoyltransferase (ZDHHC8) and Muellerian-inhibiting factor (AMH). Those overlapped genes were involved in phosphorus metabolic process, developmental processes, and responses to stimuli.
DAAM1 is a member of the formin family which modulates the nucleation of linear actin filaments. The cytoskeleton comprising microfilaments, intermediate filaments, and microtubules, is indispensable to the survival of cells. The cytoskeleton is involved in a variety of processes, including intracellular trafficking, division, migration, motility, adhesion, differentiation, and cell shape determination (Moujaber and Stochaj, 2020). The dramatic cytoskeletal-dependent phenomenon may be directly influenced by DAAM1. Cytoskeleton remodeling is fundamental to spermatogenesis and, consequently, male fertility; thus, comprehending the various proteins and mechanisms that regulate their dynamics is critically important (Li et al., 2018). Thus, DAAM1 regulates actin remodeling in the cytoplasm of germ cells throughout the differentiation phases of the seminiferous cycle and facilitates actin polymerization in type B spermatogonia nuclei, which is essential for DNA replication and cell division (Venditti et al., 2020). The Wnt signaling pathway is an evolutionarily conserved pathway that controls a variety of critical phases during embryonic development, including cell fate determination, cell polarity, cell migration, neural patterning, and organogenesis (Mezzacappa et al., 2012). It can be divided into two categories: canonical pathway and noncanonical pathway. DAAM1 is activated and functions within noncanonical Wnt signaling also referred to as the planar cell polarity (PCP) pathway, which facilitates the activation of the small GTPase Rho to modulate gastrulation movements during embryogenesis and testis development (Habas et al., 2001; Park et al., 2006), as well as organogenesis (Li et al., 2011; Bao et al., 2012; Ajima et al., 2015). PCP modulates cell motility by modifying the actin cytoskeleton (Wallingford et al., 2002; Veeman et al., 2003). Pariante et al. (2016) investigated DAAM1 during the post-natal development of the rat testis and in rat and human sperm, indicating its potential involvement in reproduction. Melatonin has been found to protects against Cadmium-induced testicular damage by regulating DAAM1 expression (Venditti et al., 2021). Melatonin improves testicular health in rats; but further research is needed to prove its efficacy in bovine to produce high-quality spermatozoa for reproduction.
COL28A1, RPL10, TRPM3, SLITRK1, ARHGEF40, SYT1, TMEM35B, and CSPG4B exhibited significant elevated expression in the RES-MEL relative to RES-CON. The COL28A1 gene encodes the collagen XXVIII protein, which is essential components of the testicular basal lamina, which plays a key role in regulating cell development and activity (Rossitto et al., 2022).
RPL10 upregulation in RES-MEL compared with RES-CON regulates cell proliferation and development. RPL10 expression levels were similar in spermatogonia and early spermatocytes, but significantly decreased in late spermatocytes and round spermatids, indicating transcriptional suppression during and after meiotic chromosomal inactivation. RPL10l compensates for RPL10 silencing during spermatogenesis (Long et al., 2017). The variable expression of RP paralogues as RPL10L is crucial for ribosomal heterogeneity in spermatogenesis (Li et al., 2022). TRPM3 and SYT1 are important for the facilitated diffusion of a calcium (Ca^2+^) ions through a transmembrane aqueous pore or channel and the cellular response to Ca^2+^ ions, respectively. Ca^2+^ signaling is a significant modulator of cell activities, particularly involving the endoplasmic reticulum (ER) (Burgoyne et al., 2015). ER-mediated Ca^2+^ signaling may be of major significance during junction turnover associated with spermatocyte translocation and sperm release in the seminiferous epithelium (Vogl et al., 2018).
Conversely, 13 genes (Receptor-type tyrosine-protein phosphatase U (PTPRU), snRNP-E, Transmembrane protein 59-like (TMEM59L), Mucin 5B, oligomeric mucus/gel-forming (MUC5B), Anaphase-promoting complex subunit 15 (ANAPC15), family with sequence similarity 221 member A (FAM221A), Testicular spindle-associated protein SHCBP1L (SHCBP1L), Progestin and adipoQ receptor family member 5 (PAQR5), Protein phosphatase 4 regulatory subunit 3C (PPP4R3C), Dystrobrevin (DTNB), long non-coding RNA (LncRNA), SH3 and multiple ankyrin repeat domains 2 (SHANK2), RIC3 acetylcholine receptor chaperone (RIC3) exhibited greater expression in the RES-CON vs. RES-MEL without any differential methylation changes. Most of these genes exhibited some functions involved in embryonic developmental abnormalities and preterm birth linked to maternal undernutrition. Moreover, most of these genes are implicated in male fetal development and spermatogenesis. Protein phosphatase 4 (PP4) is a crucial in numerous essential cellular pathways, encompassing the DNA damage response which comprises DNA repair, cell cycle regulation, and apoptosis (Park and Lee, 2020). Gene ontology analysis for testicular SHCBP1L suggested its involvement in male meiosis cytokinesis, and spermatogenesis collaborating with HSPA2 to sustain spindle integrity during meiosis in male germ cells. SHCBP1L is expressed in male germ cells in the testis, but not in female germ cells and mature sperm. Therefore, SHCBP1L is essential for germ cell division and spermatogenesis in mammals (Liu et al., 2014).
The significant DMGs of compromised pregnant dams in RES-CON relative to ADQ-CON have significant function in fetal testicular development (i.e., anatomical structure development, multicellular organism development, developmental process, cell differentiation, system development, regulation of cell morphogenesis, and catalytic activities). Interestingly, certain DMGs implicated in fetal testicular development exhibit hypomethylated (Neurogenic locus notch homolog protein 1 (NOTCH1), Rho GTPase-activating protein 7 (DLC1), Carbohydrate sulfotransferase (CHST12), A-kinase anchoring protein 13 (AKAP13), and TBC1 domain family member 22A (TBC1D22A), while others demonstrate hypermethylated (Rho guanine nucleotide exchange factor 19 (ARHGEF19), Insulin Like Growth Factor 2 Receptor (IGF2R), Transmembrane ascorbate-dependent reductase CYB561 (CYB561), Tumor necrosis factor ligand superfamily member 8 (TNFSF)8, and Peptidyl-prolyl cis-trans isomerase (PPIF). This aligns with the findings of Paradis et al. (2017), which indicate the potential consequences of insufficient maternal nutrition and the possible regulators of modified gene expression patterns, such as DNA methylation of imprinted genes like Insulin Like Growth Factor 2 (IGF2) and IGF2R, as well as microRNA expression. The murine models offer a new tool for identifying essential variables for gonocyte maintenance in the male embryonic gonad post sex determination and indicate a potential functional role for NOTCH signaling exclusively in Sertoli cells. Additionally, NOTCH signaling controls the maintenance and differentiation of Leydig progenitor cells during embryonic development (Tang et al., 2008). Research has revealed that AKAPs are crucial in regulating metabolism, growth, development, and reproduction (Ng et al., 2019; Melick et al., 2020).
The expression of KYAT1 gene was significantly increased in the ADQ-MEL relative to the ADQ-CON group without alterations in methylation for this gene. The kynurenine pathway is implicated in various activities, such as antioxidative mechanisms (Xu et al., 2018), modulation of apoptosis and endothelial dysfunction (Wang et al., 2014). These mechanisms are essential factors in placental development and vital for a successful pregnancy. Furthermore, a comparison of ADQ-MEL with ADQ-CON revealed certain genes that were hypomethylated (SCARF1, TRIO, and RBFOX3) and others that were hypermethylated (DCHS2, COLEC11, Retinoic acid receptor beta (RARB), TTLL8, and PALLD). These genes were not significantly expressed, yet they were primarily engaged in animal organ development, and the positive regulation of biological process as reproduction. Crucially, depending on the underlying genetic sequence, DNA methylation in various genomic locations may have varying effects on gene activity (Moore et al., 2013).
Lastly, five genes exhibited significant greater expression in RES-MEL relative to ADQ-MEL, including Histone type 2-B (H2B) located on chromosome 23. Additionally, two genes (PTPRU, Tudor domain containing 10 (TDRD10) exhibited significant elevated expression in ADQ-MEL vs. RES-MEL without showing any differential methylation alternations for those genes. Some hypomethylated genes (E3 ubiquitin-protein ligase LNX (LNX1), and Complement component C6 (C6), and hypermethylated genes (Paralemmin-1 (PALM), Transporter (SLC6A3), Pleckstrin homology and RhoGEF domain containing G5 (PLEKHG5), and Phosphoribosyl pyrophosphate synthase-associated protein 1 (PRPSAP1)) are involved in positive regulation of biological processes, regulation of developmental process, and regulation of responses to stimuli. In rats, the meiotic phase of spermatogenic differentiation involves the expression of the testis-specific H2B (TH2B) histone gene. There exists a substantial correlation between the expression of the TH2B gene in germ cells and DNA hypomethylation (Choi and Chae, 1991). In spermatids, protamines replace nucleosomal histones to form a more compact chromatin architecture, which is essential for enhancing sperm motility and protecting DNA during sperm maturation (Oliva, 2006; Qian et al., 2013). Besides its biological role in sperm development, differentiation, motility, nucleosome assembly, chromatin remodeling, and spermatid nucleus differentiation, TH2B is significantly correlated with gene ontology in relation to molecular processes in chromatin and DNA binding (Kutchy et al., 2017). The distribution of TH2B was varied throughout all chromosomes, excluding the sex chromosomes (Patankar et al., 2021). PTPs dephosphorylate phosphate groups from protein tyrosine residues to modulate many cellular signaling pathways via their catalytic activity. The deletion or overexpression of several PTPs adversely affects normal pregnancy and embryonic development, resulting in reproductive disorders and compromised early embryonic development (Du et al., 2023). The piRNA pathway and gametogenesis are closely linked to the majority of the 12 mammalian TDRD proteins among them TDRD10, which have either male germline-specific or germline-enriched expression patterns (Jin et al., 2009; Siomi et al., 2010). DNA methylation has been linked to gene regulation and cell differentiation, according to multiple studies (Holliday and Pugh, 1975; Compere and Palmiter, 1981).
Many of these DMGs and DEGs gene sets altered in response to melatonin supplementation are implicated in biological and developmental processes and pathways. It is now well acknowledged that one of the main epigenetic factors affecting gene activity is DNA methylation, working in tandem with other regulators. These results suggested that melatonin, a cost-effective supplement, is crucial for fetal development and beneficial regulation of biological process for compromised pregnancy in cattle. However, there exists a limitation in this study, especially within the ADQ-CON group, n = 3, hence elevating the probability of a false positive for a gene. Moreover, future research should focus on the long-term effects of these epigenetic changes on bull development and reproductive efficiency. These new findings on developmental programming’s maternal effects emphasize the need to prioritize male prenatal development.
Conclusion
This study indicates that melatonin supplementation during mid- to late-gestation positively affects the fetal testicular tissue epigenome and transcriptome. These findings suggest that male reproductive potential can be programmed, but adult testicular morphology and semen quality studies are needed. The integration of DMGs and DEGs in the omics profiles of fetal testicular investigation showed little overlap in significant expression at the time of sampling. Nonetheless, the overlapping genes identified in the DMGs and DEGs, such as the DAAM1 gene, offer a valuable opportunity for future research to investigate their relevance in the growth and maturation of the testes in male calves from birth to puberty. Epigenetics provide a new perspective on embryonic development and understanding how melatonin and/or dietary restriction affect epigenetic modifications is crucial.
Supplementary Material
skaf455_Supplementary_Data
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Ajima R. , Bisson J. A., Helt J. C., Nakaya M. A., Habas R., Tessarollo L., He X., Morrisey E. E., Yamaguchi T. P., Cohen E. D. 2015. DAAM 1 and DAAM 2 are co-required for myocardial maturation and sarcomere assembly. Dev. Biol. 408(1):126–139. 10.1016/j.ydbio.2015.10.00326526197 PMC 4765503 · doi ↗ · pubmed ↗
- 2Andrade L. P. , Rhind S. M., Rae M. T., Kyle C. E., Jowett J., Lea R. G. 2013. Maternal undernutrition does not alter sertoli cell numbers or the expression of key developmental markers in the mid-gestation ovine fetal testis. J. Negat. Results Biomed. 12:2. 10.1186/1477-5751-12-223295129 PMC 3584724 · doi ↗ · pubmed ↗
- 3Bao B. , Zhang L., Hu H., Yin S., Liang Z. 2012. Deletion of a single-copy DAAM 1 gene in congenital heart defect: a case report. BMC Med. Genet. 13:63. 10.1186/1471-2350-13-6322857009 PMC 3482563 · doi ↗ · pubmed ↗
- 4Brockus K. E. , Hart C. G., Gilfeather C. L., Fleming B. O., Lemley C. O. 2016. Dietary melatonin alters uterine artery hemodynamics in pregnant holstein heifers. Domest. Anim. Endocrinol. 55:1–10. 10.1016/j.domaniend.2015.10.00626641925 · doi ↗ · pubmed ↗
- 5Burgoyne T. , Patel S., Eden E. R. 2015. Calcium signaling at ER membrane contact sites. Biochim. Biophys. Acta. 1853(9):2012–2017. 10.1016/j.bbamcr.2015.01.022.25662816 · doi ↗ · pubmed ↗
- 6Caton J. S. , Crouse M. S., Reynolds L. P., Neville T. L., Dahlen C. R., Ward A. K., Swanson K. C. 2019. Maternal nutrition and programming of offspring energy requirements. Transl Anim Sci. 3(3):976–990. 10.1093/tas/txy 127.32704862 PMC 7200455 · doi ↗ · pubmed ↗
- 7Choi S. W. , Friso S. 2010. Epigenetics: a new bridge between nutrition and health. Adv. Nutr. 1(1):8–16. 10.3945/an.110.100422043447 PMC 3042783 · doi ↗ · pubmed ↗
- 8Choi Y. C. , Chae C. B. 1991. DNA hypomethylation and germ cell-specific expression of testis-specific H 2B histone gene. J. Biol. Chem. 266(30):20504–20511. 10.1016/S 0021-9258(18)54953-X.1718964 · doi ↗ · pubmed ↗