The emerging role of calsequestrin 2: from calcium sensor and modulator to arrhythmia driver
Humam Emad Rajha, Baha H. Abuajameia, Ali Mohamed Barhoma, Ibrahim El-Arabi Hashem, Zeyaul Islam, Christopher Lai, F. Anthony Lai, Michail Nomikos

TL;DR
Calsequestrin 2 (CASQ2) regulates calcium in heart cells and its dysfunction causes arrhythmias, with new therapies targeting CASQ2 for precision cardiology.
Contribution
This review integrates structural, functional, and pathological insights to position CASQ2 as a central target in arrhythmia and precision cardiology.
Findings
CASQ2 dysfunction is linked to arrhythmias like CPVT and sudden cardiac death.
CASQ2's structure and Ca2+ binding modulate cardiac Ca2+ dynamics and excitation-contraction coupling.
New therapies aim to stabilize CASQ2 or target its gene expression for precision treatments.
Abstract
Calsequestrin 2 (CASQ2) has emerged as a central sensor and modulator of calcium (Ca2+) dynamics in sarcoplasmic reticulum (SR), influencing both health and disease. This review explores the molecular architecture and multifunctional roles of CASQ2, beginning with its domain organization and Ca2+-binding properties and detecting how its folding and supramolecular assembly modulate Ca2+ storage and release within cardiac muscle. Post-translational modifications, genetic regulatory mechanisms and CASQ2’s multipartner interactome; including Ryanodine receptor 2 (RyR2), triadin and junctin are also discussed to highlight potential models in which complex stoichiometry and luminal Ca2+ dictate channel refractoriness and excitation-contraction coupling. Disruption of CASQ2 function is increasingly recognized as a driver of certain types of arrhythmias, notably catecholaminergic polymorphic…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Figure 1
Figure 1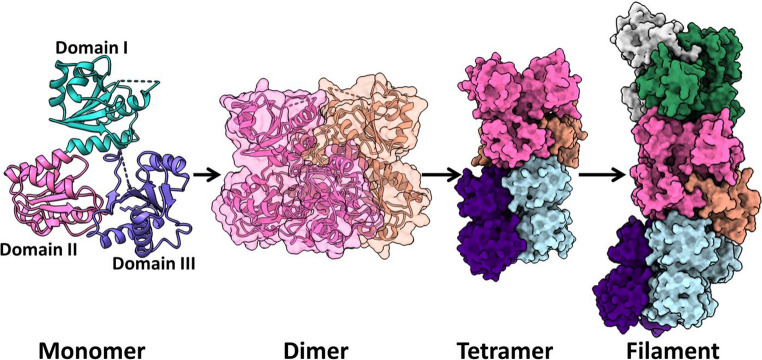 Figure 2
Figure 2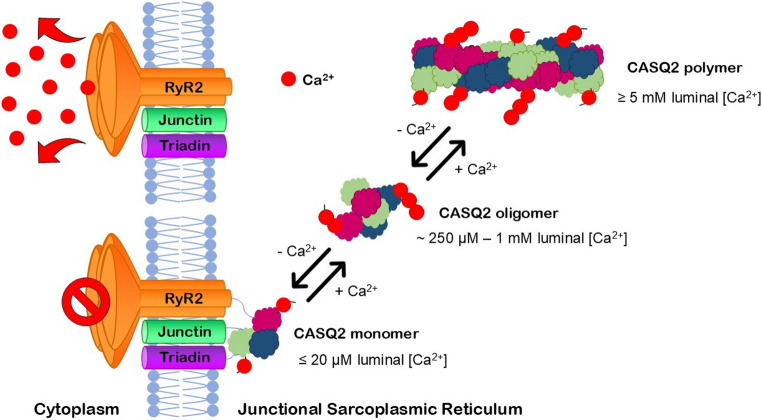 Figure 3
Figure 3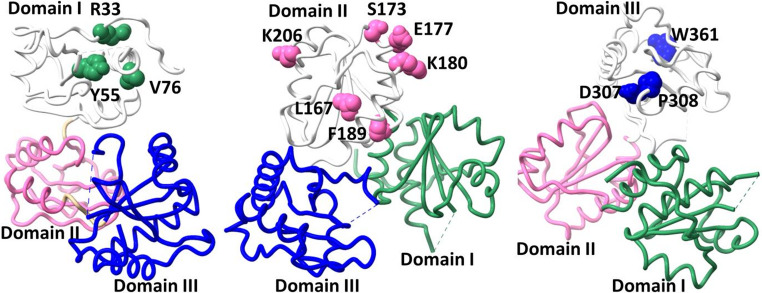 Figure 4
Figure 4- —Qatar University
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCardiac electrophysiology and arrhythmias · Ion channel regulation and function · Ion Channels and Receptors
Introduction
Few elements are as vital to human life as calcium (Ca²⁺), a mineral whose influence reaches every corner of human physiology [1]. Beyond its well-known function in fortifying bones and teeth, Ca²⁺ is crucial for critical activities in the muscles, kidneys, and the nervous system. Meanwhile, in soft tissues, Ca²⁺ serves as a messenger, regulating essential processes such as cellular signaling, blood clotting, and hormone secretion [2].
However, the most dynamic role of Ca²⁺ is possibly in muscle physiology, as fluctuations in intracellular Ca²⁺ dictate the contraction and relaxation of muscle fibers [3]. A delicate balance in intracellular Ca²⁺ levels ensures the synchronization of systole and diastole, directly affecting cardiac output [4]. Therefore, disruptions in Ca²⁺ homeostasis can trigger a cascade of pathological events, leading to arrhythmias and cardiomyopathies [5].
In muscle cells, the intricate storage and dynamic release of Ca²⁺ are tightly regulated by the sarcoplasmic reticulum (SR), a specialized intracellular organelle that serves as the primary reservoir for Ca²⁺ [6]. Within the SR, Ca²⁺-binding proteins, such as calsequestrin (CASQ), act as the guardians of this reservoir, ensuring precise control over Ca²⁺ availability. Notably, CASQ is the most abundant Ca²⁺-binding protein localized in the SR of skeletal and cardiac muscle [7].
The history of CASQ began in 1971, when MacLennan and Wong first identified CASQ in rabbit skeletal muscle tissues [8, 9]. Consequently, this discovery laid the foundation for further studies into Ca²⁺-binding proteins, and a few years later, CASQ2 was identified as the cardiac isoform of CASQ, highlighting its important role in cardiac and slow-twitch muscle fibers [10].
Vertebrates’ genome contains two closely related genes, CASQ1 and CASQ2, which exhibit notable homology but distinct expression patterns. CASQ1 is exclusively expressed in fast-twitch skeletal muscle fibers, while both CASQ1 and CASQ2 are found in slow-twitch skeletal muscle fibers. Remarkably, cardiomyocytes express only CASQ2, which is uniquely adapted to the heart’s specific needs for precise and rhythmic Ca²⁺ handling and contraction [7, 11].
As the sole CASQ isoform within cardiac muscle, CASQ2 is fundamental to Ca²⁺ buffering and regulation within the SR. Given the heart’s high demand for rapid and coordinated contraction, the precise regulation of Ca²⁺ by CASQ2 is indispensable for maintaining normal cardiac rhythm and function [12].
Forming a quaternary complex with triadin, junctin, and ryanodine receptor 2 (RyR2), CASQ2 facilitates accurate communication between luminal Ca²⁺ levels and Ca²⁺ release channels. This interaction ensures tightly regulated Ca²⁺-induced Ca²⁺ release (CICR) during cardiomyocyte excitation-contraction (EC) coupling, thereby ensuring essential rapid and synchronized contractions of cardiac muscle. Additionally, CASQ2’s structural features, including its thioredoxin-like domains and acidic C-terminal tail, allow it to dynamically respond to changes in SR Ca²⁺ levels while maintaining the balance necessary for optimal cardiac function [12].
Building on these foundational insights into CASQ2’s structure and function, further breakthroughs in the late 1990 s and early 2000 s revealed the significance of CASQ2. A notable advancement came in 1998 when the first atomic resolution crystallographic structure of CASQ2 was published, providing insights into its Ca²⁺-binding mechanism and polymeric structure [13]. Subsequently, the association of CASQ2 with catecholaminergic polymorphic ventricular tachycardia (CPVT), a form of arrhythmogenic cardiac disease, was first established in 2001 through the discovery of missense variants in a family with autosomal recessive CPVT. In the following year, nonsense variants were identified in another family with a similar condition. Consequently, this was a pivotal moment in connecting CASQ2 dysfunction to cardiac arrhythmias [14].
Following these landmark discoveries, this review aims to provide a comprehensive update on CASQ2’s central role in cardiac muscle physiology, elucidating and emphasizing its structure, function, regulation, and interactions in cardiac muscle. It delves into the pathological consequences of CASQ2 dysfunction, linking it to arrhythmogenic cardiac diseases such as CPVT. When specific studies on CASQ2 or its related diseases are not available, this review attempts to extrapolate, associate and link findings from CASQ1 or CASQ studies in general under the implicit understanding that many molecular and pathophysiological mechanisms are to a large extent conserved. Furthermore, this review also highlights recent advancements in understanding CASQ2’s role and discusses emerging potential therapeutic approaches targeting its pathways, striving to inspire further research into its function and innovative approaches to address CASQ2-associated cardiac disorders. This review positions CASQ2 as not just a protein of interest but as a cornerstone in cardiac medicine.
Structure
Structural and functional overview of CASQ2 in cardiac muscle
An essential feature of efficient Ca^2+^ handling in cardiac muscle is the rapid availability of Ca²⁺ on demand, which is supported by the highly acidic CASQ2 protein and its coordinated interaction with key components of the Ca^2+^ release unit - junctin, triadin, and the RyR2 [15–17]. Despite its modest size (~ 45 kDa), CASQ2 is the most abundantly expressed protein within the lumen of the junctional sarcoplasmic reticulum (jSR), with concentrations reaching up to 100 mg/mL [9].
The CASQ2 gene is situated on chromosome 1, at locus p13.3-p11, and is made up of 11 exons [18, 19]. The translated human CASQ2 protein comprises 399 amino acids. However, the initial 19 amino acids form a signal peptide sequence that is likely removed before CASQ2 reaches its final location—the jSR [20–22]. Although the length of the full CASQ2 protein may differ between species, there is a high level of similarity (approximately 89–96%) that is maintained among mammalian species [23].
Domain organization and Ca2+-binding properties
The quaternary structure of the compact, globular CASQ2 protein is composed of five distinct regions (Fig. 1): a brief N-terminal segment (amino-terminal loop), three acidic thioredoxin-like (TrxL) domains (referred to as domains I, II, and III), and an unstructured, flexible, and acidic C-terminal tail [21]. This architecture is reminiscent of that found in the endoplasmic reticulum (ER) luminal oxidoreductases [21]. The large number of acidic residues are distributed on the surface, particularly within loops and turns of the structure, facilitating high-capacity Ca^2+^-binding. Thus, maintaining the structural integrity of each region is essential for CASQ2’s ability to bind Ca²⁺ and to undergo polymerization given that the tri-domain structure allows for flexibility and extensive surface area necessary for CASQ2’s role in Ca^2+^ buffering [24].Fig. 1. Domain organization of calsequestrin 2 (CASQ2). Structural architecture of the CASQ2 protein, which is composed of three distinct acidic thioredoxin-like (TrxL) domains (Domain I, Domain II, Domain III) and an unstructured C-terminal tail. The domains are arranged in a compact, globular structure, with each domain characterized by a β-sheet flanked by α-helices. The figure highlights the flexibility of CASQ2, enabling it to bind Ca^2+^ and undergo polymerization, which is critical for Ca^2+^ buffering in cardiac muscle cells. The N-terminal and C-terminal regions, marked by their acidic residues, are key in the protein’s functionality, contributing to its role in Ca^2+^ storage and release within the jSR
The three TrxL domains (which make the overall structure of human CASQ2) are nearly identical in architecture. Each domain is composed of approximately 100 amino acids and features a five-stranded β-sheet flanked on both sides by two α-helices [21]. These three domains adopt a globular conformation, each exhibiting the conserved architecture of a hydrophobic core surrounded by β-sheets and α-helices. The domains span the following residue ranges: domain I (residues 22–142), domain II (143–246), and domain III (247–370). The conserved hydrophobic core and surface-exposed acidic residues contribute to the generation of the strong electronegative potential on the protein surfaces [21].
Several structural studies using crystallography, size-exclusion chromatography, cross-linking and light scattering have highlighted the Ca^2+^-dependent dynamic polymers [25–27]. CASQ2 exists in multiple conformational states resulting in self-assembled higher oligomeric forms including filaments (Fig. 2). Ca^2+^-dependent transition from monomer to dimer has been reported which eventually led to inter-dimer interaction and higher-order multimer [28, 29]. The dimer stacks along a screw axis to create a filament where each dimer is rotated 90° relative to its neighbours. In the filament, CASQ2 structure exhibits a helical architecture with outer and inner helices formed by different thioredoxin domains [29].Fig. 2. Calsequestrin 2 (CASQ2) higher order multimerization into filaments. Structure of several conformational states of CASQ2. The protein exists as a monomer and a wide range of higher order multimers. Cartoon as well as surface representation of CASQ2 monomer, dimer, tetramer and filament (PDBID: 6OWV). The CASQ2 monomer consists of three thioredoxin domains (domain I, II and III represented in green, pink and blue respectively). The higher multimer forming contacts exist between dimers and stacks along the screw axis to form the filament
The full functional roles of the TrxL domains are not yet fully understood. However, due to the presence of this conserved protein fold, both CASQ1 and CASQ2 are grouped within a large TrxL protein superfamily comprising over 4,000 members, all characterized by the presence of a TrxL domain. The TrxL superfamily also encompasses the protein disulfide isomerase (PDI) family [30, 31]. Despite their conserved tertiary structure, the proteins within the superfamily exhibit less than 30% similarity at the primary sequence level. Only a few of the proteins in this superfamily have been functionally characterized [30, 31].
Both CASQ isoforms lack the canonical C-x-x-C (cysteine-x-x-cysteine) catalytic motif typically required for thioredoxin enzymatic function [31]. Notably, the N-terminus of CASQ2—but not CASQ1—contains two highly conserved cysteine residues. However, their locations suggest they are unlikely to contribute to isomerase activity [30, 31]. Cys17 is part of the signal peptide, which is cleaved during protein synthesis and thus absent in the mature protein. Cys53 is located deep within the hydrophobic core of domain I’s thioredoxin-like fold, rendering it inaccessible for interaction with substrate proteins [30, 32].
The C-terminal domain of CASQ, rich in aspartic and glutamic acid residues, serves as the primary Ca^2+^-binding region on the monomer [33]. An extended C-terminal end contains more than 70% of acidic residues. Therefore, removing this domain from CASQ proteins leads to a reduction of over 50% in their Ca^2+^-binding capacity [33]. Notably, CASQ2 has a longer, more negatively charged C-terminus than CASQ1, contributing to the protein’s solubility and Ca^2+^-binding capacity as well as anchoring it to the jSR membrane [34].
Ca2+-dependent folding, polymerization and supramolecular organization
CASQ2 has a dynamic and reversible structure; at low Ca^2+^ levels, CASQ2 exists in a molten globule state, then folds into compact monomers as Ca²⁺ rises [13, 16]. These monomers dimerize through N-terminal (front-to-front) interactions and further polymerize via C-terminal (back-to-back) stacking [24, 35]. Polymerization enhances CASQ2’s Ca^2+^-buffering capacity by forming negatively charged pockets that attract and bind Ca²⁺ ions [21, 36].
As for CASQ’s secondary and tertiary folding which is maintained by cations, all CASQ isoforms are rich in carboxylate groups from the aspartic and glutamic acid residues, giving human skeletal and cardiac CASQ isoelectric points of 4.0 and 4.2, respectively. At low ionic strength (below 100 mM KCl), electrostatic repulsion between these negative charges causes the protein to adopt an extended, random coil conformation [37, 38]. Multiple, monovalent, or divalent cations also play a role in directing the folding of CASQ’s highly similar, negatively charged TrxL domains where several hydrophobic interactions support the interior of the domains [39, 40]. The lowest ionic concentration necessary for maintaining CASQ’s secondary and tertiary folding depends on the coordination number and ionic radius of the cation. Among various cations, Ca²⁺ is the most efficient, showing effective binding at low concentrations and exhibiting notable cooperativity even under modest ionic conditions [41, 42].
In a study by Titus et al. (2020) to examine CASQ2’s filament structure, domain I forms an outer helical collar, while domains II and III form a compact inner double helix. The filament is built from stacked dimers rotated 90°, forming a left-handed helix with a continuous, electronegative lumen. This arrangement and binding site mapping were revealed under low pH crystallization conditions, enabling visualization of biologically relevant multimeric assembly for the first time [36].
Function
CASQ2 has a variety of primary functions which include Ca²⁺ storage and buffering, regulation of Ca²⁺ release, EC coupling, maintenance of Ca²⁺ homeostasis, and stress response modulation. These roles emphasize its importance in cardiac function and its association with various pathophysiological conditions.
Ca²⁺ storage and buffering
CASQ2 plays a critical role in Ca²⁺ storage and buffering within the SR [43]. It binds approximately 40–60 moles of Ca²⁺ per mole of protein. CASQ2’s low affinity binding enables the SR to maintain a substantial reservoir of Ca²⁺ for rapid mobilization during muscle contraction [24]. At high luminal Ca²⁺ levels (> 1 mM), CASQ2 polymerizes into filamentous structures, enhancing its Ca²⁺-binding capacity [24]. Conversely, at low Ca²⁺ levels, it depolymerizes, enabling the bound Ca²⁺ to be released into the cytosol to initiate contraction [24, 44].
As a reversible Ca^2+^ buffer, CASQ2 reduces free Ca²⁺ concentration within the SR, lowering the gradient against which the sarcoplasmic reticulum Ca²⁺-ATPase (SERCA) operates [45, 46]. This buffering action supports efficient Ca²⁺ reuptake and prevents cytotoxicity caused by elevated Ca²⁺ levels [47]. Dysregulation of CASQ2’s buffering capacity, observed in pathological conditions such as heart failure, leads to impaired Ca^2+^ homeostasis, contributing to arrhythmias and contractile dysfunction [48, 49].
Regulation of Ca²⁺ release
CASQ2 directly interacts with RyRs, stabilizing these SR Ca²⁺ release channels and modulating their responsiveness to Ca²⁺. It acts as a sensor for luminal Ca²⁺ levels, promoting RyR opening at high Ca²⁺ concentrations and inhibiting it at low concentrations [50, 51]. This precise regulation ensures controlled Ca²⁺ dynamics during muscle contraction [52].
During CICR, a small influx of Ca²⁺ through voltage-gated Ca²⁺ channels activates RyRs [50, 53]. CASQ2 undergoes conformational changes upon Ca²⁺ binding, enhancing RyR opening and amplifying Ca²⁺ release from the SR [52]. This rapid increase in cytosolic Ca²⁺ triggers actin-myosin interactions, driving contraction. Mutations in CASQ2, such as R33Q, disrupt this interaction, leading to spontaneous Ca²⁺ release and arrhythmias, such as catecholaminergic polymorphic ventricular tachycardia (CPVT), particularly under stress [54–56].
Excitation-Contraction (EC) coupling
In cardiac EC coupling, an action potential spreads over the sarcolemma, penetrates the T-tubules and causes activation of voltage-gated L-type Ca²⁺ channels (dihydropyridine receptors) [12]. The ensuing influx of a small amount of extracellular Ca²⁺ into the cytosol, which binds to the cytosolic region of RyR2 [57], serves as the spark for the massive release of Ca²⁺ from the SR through RyR2 channels, a process known as CICR [58].
CASQ2 acts as a high-capacity, moderate-affinity Ca²⁺ buffer within the SR lumen. It binds large amounts of Ca²⁺, thus maintaining a readily releasable pool of Ca²⁺ for subsequent contractions. By doing so, CASQ2 sustains the Ca²⁺ gradient across the SR membrane and helps ensure that sufficient Ca²⁺ is available when needed for contraction.
In addition to regulating release, CASQ2 supports efficient Ca²⁺ reuptake during muscle relaxation. As it buffers Ca²⁺ in the SR, it facilitates the activity of the SERCA pump, which transfers cytosolic Ca²⁺ back into the SR. By doing so, CASQ2 helps restore SR Ca²⁺ levels between heartbeats and prevents cytosolic Ca²⁺ overload, a condition that could otherwise promote delayed afterdepolarizations and trigger arrhythmias [59–61].
This finely tuned balance is especially crucial during conditions of increased cardiac demand, such as exercise or emotional stress, where heart rate and intracellular Ca²⁺ fluxes are elevated [4]. Under such circumstances, CASQ2’s role in stabilizing SR Ca²⁺ content becomes even more important for preserving the synchrony between electrical excitation and mechanical contraction.
Maintenance of Ca²⁺ homeostasis
CASQ2 is an example of specialized evolutionary adaptation optimized for CICR in cardiac muscle [62]. Unlike CASQ1, which supports depolarization-induced Ca²⁺ release in skeletal muscle, CASQ2’s function ensures precise regulation of Ca²⁺ signaling dynamics in response to electrical activity [60]. It balances free and bound Ca²⁺ within the SR, maintaining free Ca²⁺ at approximately 1 mM while allowing total concentrations to reach up to 19 mM [51, 63]. This balancing act is crucial for Ca²⁺ reuptake and availability during contraction [64].
CASQ2 also modulates store-operated Ca²⁺ entry (SOCE), a pathway for replenishing SR Ca²⁺ stores post-depletion [65]. It interacts with stromal interaction molecule 1 (STIM1), an SR Ca²⁺ sensor, inhibiting SOCE through luminal Ca²⁺ stabilization. Loss of CASQ2 enhances SOCE activity, disrupting Ca²⁺ homeostasis and increasing susceptibility to arrhythmias [51, 66, 67].
In addition to Ca²⁺ binding, CASQ2 exhibits ion selectivity, interacting with divalent and monovalent cations [46]. While its physiological relevance is dominated by Ca²⁺ binding, interactions with other ions, such as cadmium or zinc, may influence its structure and function under pathological conditions [68].
Stress response and protection
CASQ2 contributes significantly to the cardiac response to physiological and pathological stress. It interacts with the unfolded protein response (UPR) sensor Inositol-Requiring Enzyme 1 alpha (IRE1α), preventing its dimerization and subsequent UPR activation under normal conditions [60, 69]. Disruption of this interaction, as seen in CASQ2 mutations, exacerbates stress signaling pathways, leading to cardiomyopathy and arrhythmias [70]. By modulating stress responses, CASQ2 ensures efficient cardiac function even under adverse conditions [71].
CASQ2 also protects cardiac tissue from oxidative stress by minimizing Ca²⁺ overload-induced damage [72]. Pharmacological agents such as dantrolene, which enhance CASQ2 polymerization and buffering capacity, underscore its potential as a therapeutic target in stress-induced cardiac conditions [73]. Additionally, CASQ2’s role in maintaining rhythm stability by preventing spontaneous Ca²⁺ release is vital, particularly during elevated adrenergic stimulation [74]. Evidence from knockout animal models links CASQ2 dysfunction to sinoatrial node defects and arrhythmias such as atrial fibrillation (AF) [75].
Developmental role
CASQ2 may also play a role in cardiac and skeletal muscle development. Its expression, regulated by transcription factors like MEF-2 C and pathways such as calcineurin/Nuclear Factor of Activated T-cells (NFAT), suggests its involvement in stabilizing Ca²⁺ signaling mechanisms during development [76–78].
CASQ2 knockout animals exhibit increased susceptibility to stress-induced cardiac dysfunction, highlighting the importance of CASQ2 in maintaining Ca²⁺ homeostasis during early development. Disrupted CASQ2 expression can impair myocyte maturation and predispose the heart to arrhythmias or cardiomyopathy, underscoring its role in establishing a stable Ca²⁺ environment during growth [79, 80].
Regulation
Phosphorylation
Phosphorylation of calsequestrin 2 plays an important role in its function and structural dynamics. CASQ2 gets phosphorylated on its C-tail by casein kinase 2 [12]. This enzyme acts on specific sites for phosphorylation, such as Ser385 and Ser393. These sites were identified as phosphorylation sites of CASQ2 by Park et al. (2005) using mass spectrometry-based phosphoproteomics [81]. Functionally, dual phosphorylation of these sites significantly enhance CASQ2 helical content, solubility, and Ca²⁺-binding capacity when Ca²⁺ levels exceeded 6 mM. Structural studies highlighted that these modifications alter the conformation and charge of CASQ2’s C-tail, modulating an important role in CASQ2 function. In contrast, phosphorylation of one site has no significant effect, highlighting the crucial role of dual site phosphorylation in modulating CASQ2 function [81].
Newly synthesized CASQ2 is fully phosphorylated; suggesting that phosphorylation is essential for its initial structural integrity and functionality. Furthermore, dephosphorylation of CASQ2 in cardiac tissue has been documented with regard to a constitutive phosphate turnover of CASQ2 in the heart, and this turnover may be associated with its intracellular transport and SR localization [82]. While direct functional data linking CASQ2 dephosphorylation to Ca²⁺-release during cardiac EC coupling remain limited, this finding emphasizes that a dynamic balance between phosphorylation and dephosphorylation is likely vital for CASQ2 structural integrity and function.
Phosphorylation of CASQ2 has been also shown to play a role in its protein transport and localization within the cell [12] by making CASQ2 highly soluble and compact. However, McFarland and colleagues (2010) found that the loss of CASQ2 phosphorylation enhances anterograde trafficking [83]. The co-translocational hypothesis resolves this discrepancy in the literature, suggesting that phosphorylation takes place in the cytoplasmic domain of CASQ2 during its translocation through the translocon into the ER, before it fully localizes to the lumen of the SR.
Another critical yet less-studied kinase involved in CASQ2 phosphorylation is Fam20C. Fam20C phosphorylates CASQ2 and other SR proteins, playing an essential role in regulating Ca²⁺ homeostasis and providing cardioprotection by preventing heart failure. Unlike casein kinase 2, which targets specific C-tail residues, Fam20C has broader regulatory effects on SR protein function, contributing to overall Ca²⁺ dynamics [84].
Glycosylation
In addition to phosphorylation, another essential post-translational modification that calsequestrin undergoes is N-linked glycosylation. This modification significantly influences its structure, stability, and function [23]. This process occurs co-translationally in the ER and involves the enzymatic activity of the oligo-saccharyl transferase complex [85]. This enzyme transfers a pre-assembled glycan moiety to specific asparagine residues within the consensus motif N-X-S/T of CASQ during its synthesis and translocation into the ER lumen. Human CASQ2 has two known glycosylation sites, N212 and N335 [85]. Glycosylation at these sites stabilizes the tertiary structure of CASQ, particularly Domain III [86], which plays a vital role in monomer alignment during polymerization. The presence of oligosaccharides acts as a “molecular guiderail”, ensuring directing or stabilizing the proper formation and alignment, and the generation of stable and linear CASQ polymers [87].
Glycosylation not only enhances the solubility and Ca²⁺-binding efficiency of CASQ, it also enables it to respond effectively to changes in Ca²⁺ concentrations during EC coupling [88]. Furthermore, glycosylation promotes CASQ trafficking through the secretory pathway to its functional site at the SR junction (SRJ). Though this observation was originally reported for the skeletal muscle isoform CASQ1 [88], cardiac CASQ2 exhibits more extensive glycosylation, with CASQ2 having an average of six mannoses per glycosylation site compared to one mannose in CASQ1, reflecting its unique functional demands in cardiac tissue [23]. Disruptions in glycosylation, whether due to mutations or enzymatic deficiencies, can impair CASQ’s polymerization and Ca²⁺-buffering capacity, contributing to pathological conditions such as catecholaminergic polymorphic ventricular tachycardia (CPVT) [12].
Genetic regulation
The transcription of the CASQ2 gene, encoding the cardiac-specific isoform of calsequestrin, is tightly regulated by a combination of transcription factors, promoter architecture, and Ca^2+^-dependent signaling pathways. The Ca²⁺-calcineurin/NFAT pathway plays a central role in modulating CASQ2 expression in cardiomyocytes, where NFAT acts as a positive regulator - inhibition of NFAT dephosphorylation reduces CASQ2 mRNA levels [89]. Additionally, Myocyte Enhancer Factor-2 (MEF-2) binding sites within the CASQ2 promoter are essential for transcriptional activation, and NFAT cooperates with MEF-2 to enhance gene expression [90]. This synergy highlights a Ca^2+^-sensitive mechanism where calcineurin-NFAT signaling, triggered by intracellular Ca²⁺, directly links cardiac activity to gene regulation.
The proximal promoter region of CASQ2, spanning approximately 180 base pairs upstream of the transcription start site, contains several evolutionarily conserved regulatory elements. These include a TATA box for transcription initiation, a CArG (CC(A/T)6GG) box (recognized by serum response factor (SRF) to drive muscle-specific expression), an E-box (targeted by myogenic regulators like MyoD and myogenin), and MEF-2 sites critical for muscle gene activation [91]. These motifs collectively ensure tissue-specific and developmentally appropriate CASQ2 expression, particularly in cardiac and slow skeletal muscle fibers. The interplay between SRF, MEF-2, and myogenic factors underscores the combinatorial control of CASQ2 transcription, fine-tuning its levels to match functional demands.
Beyond transcriptional regulation, hormonal and epigenetic mechanisms may further influence CASQ2 expression. Thyroid hormone has been shown to modulate CASQ isoforms levels in non-mammalian models, suggesting potential conserved roles in vertebrates [92]. Epigenetic modifications, such as DNA methylation or histone acetylation, could also contribute to silencing CASQ2 in non-muscle tissues, though experimental validation in mammals is still needed. Together, these regulatory layers - transcription factor networks, promoter architecture, Ca^2+^ signaling, and secondary hormonal/epigenetic inputs - ensure precise CASQ2 expression, optimizing Ca^2+^ handling and contractile function in the heart.
Protein interactions
Interaction with ryanodine receptor 2
Cardiac CASQ2 interacts directly with the luminal domain of RyR2 at its first luminal loop, forming a crucial component of the SR Ca²⁺ sensor [11]. Structural studies reveal that this binding occurs through electrostatic interactions between acidic residues in CASQ2’s C-terminal dimerization domain and basic residues in RyR2’s luminal loop. This interaction, supported by accessory proteins triadin and junctin, modulates RyR2 function and SR Ca²⁺ release [93]. Together, these proteins form the " Ca^2+^- release complex”, where triadin and junctin act as molecular scaffolds that enhance the physical and functional coupling between CASQ2 and RyR2.
CASQ2 serves as both a Ca²⁺ storage site and a luminal Ca²⁺ sensor, inhibiting RyR2 opening at low luminal Ca²⁺ concentrations and regulating Ca²⁺ release. At low Ca²⁺ levels (< 1 mM), monomeric CASQ2 binds tightly to RyR2, suppressing channel activity to prevent diastolic Ca²⁺ leaks. The CASQ2–RyR2 association is Ca²⁺-dependent; monomeric CASQ2 at low luminal Ca²⁺ inhibits RyR2, whereas polymerization at higher Ca²⁺ (> 5 mM) diminishes this inhibition [45]. This polymerization triggers conformational changes that reduce CASQ2’s affinity for RyR2, effectively releasing inhibition and priming the channel for systolic Ca²⁺ release. Figure 3 illustrates how CASQ2 regulates Ca^2+^ release through its interaction with RyR2.Fig. 3Ca^2+^-Dependent Regulation of RyR2 by CASQ2 within the Junctional Sarcoplasmic Reticulum (jSR) of Cardiac Muscle. At high luminal Ca²⁺ concentrations (≥ 5 mM), CASQ2 has the lowest affinity for RyR2, polymerizes in the SR lumen enhancing Ca²⁺ storage and has no inhibitory effect on RyR2. As luminal Ca²⁺ decreases (~ 250 µM − 1 mM), CASQ2 depolymerizes into small oligomers with intermediate affinity for RyR2 and minimal regulatory effect on the Ca²⁺ channel. When Ca²⁺ levels fall to ≤ 20 µM, monomeric CASQ2 has the highest affinity for the RyR2 and effectively inhibits RyR2 channel activity via interactions with triadin and junctin, preventing spontaneous Ca²⁺ release. This reversible mechanism ensures controlled Ca²⁺ dynamics critical for cardiac contraction and relaxation
Recent cryo-EM studies have mapped the precise binding interface between CASQ2 and RyR2, showing that disruption of this interaction leads to aberrant Ca²⁺ sparks and arrhythmias in cardiomyocytes [11]. Furthermore, mutations in either CASQ2 or RyR2 that impair their co-binding are linked to CPVT, underscoring the physiological importance of this interaction [94].
CASQ2 interaction with triadin
CASQ2 and triadin are key components of the junctional SR complex, critical for Ca²⁺ release regulation. Triadin is a transmembrane protein that connects CASQ2 to RyR2, forming a structural scaffold that stabilizes the Ca²⁺ release unit [60]. CASQ2 modulates RyR2 activity by inhibiting premature Ca²⁺ release through its association with triadin [7]. Disrupting this interaction increases the sensitivity of SR Ca²⁺ release to triggers, causing spontaneous Ca²⁺ oscillations and arrhythmic membrane depolarizations. The interaction also supports SR morphology; triadin ablation reduces junctional SR extent by 50%, impairing Ca²⁺ release dynamics [60]. Isoform-specific effects further refine regulation, with triadin-1 predominating in cardiac tissue and exhibiting distinct roles in RyR2 gating. Loss of triadin disrupts CASQ2-RyR2 coupling, increasing susceptibility to stress-induced arrhythmias. The binding between CASQ2 and triadin is mediated by specific amino acid motifs, triadin contains Lysine (K)–Glutamate (E)–Lysine (K)–Glutamate (E) (KEKE) motifs in its luminal domain, which are responsible for binding to CASQ2. This interaction is essential for anchoring CASQ2 to the junctional face membrane, allowing it to sequester Ca²⁺ in the vicinity of the RyR2 during Ca²⁺ uptake and release [50]. The aspartate-rich region of CASQ2 also plays a role in this interaction, providing a direct binding site for both luminal Ca²⁺ and triadin [95].
CASQ2 interaction with junctin
Junctin is an integral component of the SR Ca²⁺ release complex, directly interacting with CASQ2, triadin and RyR2 [60]. Like triadin, the junctin luminal domain is rich in charged amino acids and KEKE motifs, and this domain binds CASQ2 and stabilizes the quaternary complex [96]. Junctin facilitates the localization of CASQ2 to the junctional SR, ensuring its proper association with RyR2 for efficient Ca²⁺ release regulation [50]. CASQ2’s Ca²⁺-dependent polymerization and depolymerization cycles require junctin, highlighting its role in maintaining CASQ2’s scaffold-like structure. The CASQ2-junctin interaction is essential for modulating RyR2 gating and buffering SR Ca²⁺ [45]. Overexpression of junctin disrupts Ca²⁺ signaling by altering the organization of Ca²⁺ release units, while mutations affecting junctin’s stability impair Ca²⁺ handling and contribute to cardiac hypertrophy and contractile dysfunction.
CASQ2 and its synergistic role in the sarcoplasmic reticulum protein complex
CASQ2, along with triadin and junctin, forms a quaternary complex with RyR2. This complex is necessary for the normal operation of Ca²⁺ release in cardiac muscle. The KEKE motifs in triadin and junctin facilitate the binding of CASQ2 to RyR2, stabilizing the complex and ensuring efficient Ca²⁺ signaling [96]. The complex functions as an integrated system to facilitate Ca²⁺ storage and release in response to fluctuating luminal Ca²⁺ levels. At low SR Ca²⁺, CASQ2 remains monomeric and exerts an inhibitory effect on RyR2 via triadin/junctin. As Ca²⁺ levels rise, CASQ2 polymerizes into a filamentous matrix, weakening its interaction with RyR2 and relieving channel inhibition to permit Ca²⁺ release [51]. Disruption in this synergistic network, whether through mutations in CASQ2, triadin deficiencies, or junctin imbalances, leads to impaired Ca²⁺ homeostasis, triggering arrhythmogenic disorders and contractile dysfunction. These interactions demonstrate the delicate balance required for precise Ca²⁺ regulation in cardiac muscle [15, 93].
Pathology
CASQ2 is undoubtedly a key protein in muscular functions; without it, Ca²⁺ handling and coordination would be impaired, leading to severe pathological consequences. Any dysregulation in CASQ2 normal functioning often leads to arrhythmogenic events.
Cardiac arrhythmias are disturbances in the normal rhythm of the heart, arising from abnormalities in impulse generation or conduction. They can be broadly classified into acquired and inherited types. Acquired arrhythmias are typically caused by external factors like drugs or underlying diseases [97–101], while inherited arrhythmias result from genetic mutations. Examples of inherited arrhythmias include CPVT and arrhythmogenic right ventricular cardiomyopathy (ARVC), while long QT syndrome can be either inherited or acquired. These conditions are frequently linked to mutations in SR proteins [102–104], leading to Ca²⁺ dysregulation that manifests through early afterdepolarizations (EADs), delayed afterdepolarizations (DADs), or reentrant circuits [105].
The dysfunction of CASQ2 is associated with various inherited cardiac conditions, most notably arrhythmias. This section explores the conditions linked to CASQ2 dysfunction, highlighting the underlying mutations and pathophysiological mechanisms.
Catecholaminergic polymorphic ventricular tachycardia (CPVT)
The primary cardiac condition related to CASQ2 dysfunction is CPVT. The true prevalence of CPVT in the population is not known. An estimate of CPVT prevalence is 1:10,000 or less [106], with CASQ2-related CPVT2 accounting for about 5% of these cases [107]. Although much of the epidemiological and clinical data available in the literature describe CPVT in general, the underlying arrhythmogenic mechanism is shared across subtypes, namely, stress-induced SR Ca²⁺ leak leading to DADs.
Mutations such as R33Q, R35W, and E202K in the CASQ2 gene are commonly implicated in this CPVT2 subtype. CASQ2 mutations are often autosomal recessive and can manifest as missense, nonsense, or frameshift variants, leading to loss of CASQ2 protein functionality or stability [108]. The D307H and R33Q mutations are particularly well-documented, with studies demonstrating their role in impairing Ca²⁺-binding capacity and promoting protein misfolding [56, 79, 109]. These genetic defects reduce CASQ2’s ability to buffer Ca²⁺ ions in the SR, resulting in abnormal Ca²⁺ cycling, which is central to the pathophysiology of CPVT [110]. In individuals with CPVT, adrenergic stimulation, typically triggered by physical exertion or emotional stress [106] leads to excessive Ca²⁺ release from the SR. This abnormal release results from impaired CASQ2 function, causing DADs that can precipitate life-threatening arrhythmias, including bidirectional or polymorphic ventricular tachycardia [107, 111–113]. The compromised Ca²⁺ buffering capacity increases SR Ca²⁺ leak, further elevating cytosolic Ca²⁺ levels and heightening the risk of arrhythmogenic events.
Risk of sudden cardiac death (SCD)
Untreated CPVT is highly lethal, with approximately 30% of affected individuals experiencing at least one cardiac arrest (CA), and up to 80% suffering recurrent syncope episodes [106]. Sudden death (SD) can be the initial manifestation, especially in children [114]. Patients presenting as the index case in a family (first diagnosed) generally have a more severe phenotype and are at a higher risk of adverse events, including SCD. Even after initiating medical treatment, high-risk ventricular arrhythmia persists in most patients, with fatal events occurring in 20.6% over 7.4 years, primarily due to non-compliance with exercise restrictions or medication [115]. Long-term follow-up studies show a high prevalence of cardiac events despite beta-blocker therapy, with 75% of patients experiencing syncope and 83% requiring implantable cardiac defibrillators [116]. More mutations and their associated diseases can be found in Table 1.
Mutations in CASQ2 are highly lethal and several mutations disrupt filamentation [36]. Mapping of disease-causing point mutations onto the CASQ2 structure and its interfaces highlighted that the disease pathology is associated with defect in multimerization. The point mutations are distributed throughout the protein, and shared by three identically folded TrxL domains (domain I, domain II and domain III) (Fig. 4).Fig. 4. Location of disease associated mutations on the CASQ2 structure. CASQ2 monomeric crystal structure (PDBID: 6OWV) displaying the position of point mutations mentioned in Table 1. The locations of point mutations on CASQ2 domain I (left) are represented as spheres in green color. The locations of point mutations on CASQ2 domain II (middle) are shown as spheres in pink. Positions of domain III (right) mutations are shown as spheres in blue color. CASQ2 protein is colored domain-wise, domain I as green, domain II as pink and domain III as blue
Table 1. Distinct CASQ2 mutations, pathologies they present in, and how they May lead to these pathologiesMost Common Associated Disease(s)/Phenotype(s)Mutation: Amino Acid ChangeMutation:Nucleotide Change (cDNA Change)Mutation:Type, Domain, HomozygosityProposed Underlying Mechanisms of Ca^2+^ Dysregulation/Functional ImplicationClinical Presentation/Notes/SeverityCatecholaminergic polymorphic ventricular tachycardia (CPVT)D307H1038 G > C [117, 118]Missense, III,Homozygouswhile heterozygous carrier shows no symptoms [21, 119, 120]Leads to either reduced SR Ca²⁺ content and impaired Ca²⁺ transients, or unaltered SR content with increased Ca²⁺ leak under both resting and stimulated conditions. CASQ2 expression is drastically reduced (~ 95%), and the mutant protein exhibits impaired Ca²⁺ buffering and binding, defective polymerization, and increased susceptibility to degradation. Although properly localized to the junctional SR, the mutant CASQ2 shows reduced interaction with triadin and junctin, and a loss of both low-affinity Ca²⁺ binding and Ca²⁺ selectivity—sometimes showing aggregation in the presence of Mg²⁺. Structural changes include protein misfolding and the loss of Ca²⁺-concentration-dependent conformational shifts. These alterations culminate in decreased SR Ca²⁺ storage capacity, impaired release, and the generation of spontaneous Ca²⁺ sparks [21, 42, 108, 117, 119–123]Early onset during childhood and elevated mortality if left untreated, accompanied by resting bradycardia and a slight QTc interval prolongation [42, 108, 117, 119–123]R33Q98 G > A [124]Missense, N-terminus, Homozygous [21, 56, 125]Exhibits ~ 50% reduced expression, impaired inhibition of RyR2, and increased RyR2 sensitivity to luminal Ca²⁺, leading to higher CICR gain, more frequent Ca²⁺ sparks and waves, and decreased SR Ca²⁺ content, despite unaltered triadin binding. Some variants show enhanced buffering, but with diminished Ca²⁺-induced conformational changes, reduced polymerization, and lower Ca²⁺ binding capacity at high concentrations, despite normal Ca²⁺ affinity. These defects are further exacerbated by increased protein degradation[21, 124–127]Arrhythmia, non-sustained VT during exercise since young age which can lead to exercise-induced syncopal episodes [124]R33X97 C > T [118]Nonsense, N-terminus, Heterozygous [118, 128]Introduces a premature stop codon, resulting in absence of full-length CASQ2 and a marked reduction in functional protein levels. This leads to disrupted Ca²⁺-dependent polymerization, with aggregation at low Ca²⁺ concentrations and impaired structural integrity. Despite correct localization to the junctional SR, the mutant protein fails to fully inhibit RyR2 at low luminal Ca²⁺, contributing to reduced SR Ca²⁺ content and the emergence of spontaneous Ca²⁺ waves and sparks [118, 128, 129]Exercise-induced syncope, arrhythmias, and borderline QTc prolongation since childhood [118]L23fs + 14X62delA [118]Deletion, N-terminus, Homozygous while heterozygous siblings show no symptoms [118, 125]The frameshift deletion causes a premature stop codon 14 amino acids downstream which results in truncated CASQ2 proteins that lack Ca²⁺-binding ability [118, 125]Severe CPVT andexercise-induced syncope since young age [118]L167H500 T > A [130]Missense, II, Compound heterozygous with G112 + 5X [42, 108, 130]Increases sensitivity to proteolytic cleavage and reduces Ca²⁺-dependent polymerization. While Ca²⁺-binding affinity is preserved, binding capacity declines at high Ca²⁺ levels, and the protein aggregates at low Ca²⁺. It also loses Ca²⁺-induced conformational changes, impairing RyR2 activation at 1 mM Ca²⁺. These defects lead to reduced SR Ca²⁺ content, diminished SR Ca²⁺ release, and fewer spontaneous Ca²⁺ sparks [21, 42, 108, 129, 130]Severe CPVT with several runs of polymorphic ventricular tachycardia andmultiple syncopal events from young age [130]Y55C164 A > G [131]Missense, I, Compound heterozygous with P308L [125, 131]Loss of proper dimerization and filamentation capabilities [125, 131]Non-sustained VT under adrenaline stress; requires compound heterozygosity with P308L to manifest full CPVT phenotype [131]V76M226G > A [132]Missense SNP, I,Heterozygous [132, 133]Disrupts protein stability and polymerization, leading to reduced expression levels. It also partially impairs Ca²⁺ binding capacity [132, 133]Shows no consistent clinical presentation and is considered a rare, likely benign polymorphism found in Finnish families & Asian population [132, 133]Sudden Death (SD)E177Q529G > C [132]Missense, II,Heterozygous mutation [132]Suggested disruption of Ca²⁺ binding capacity due to substitution of a negatively charged glutamic acid with a neutral glutamine which may impair normal chelation function. However, no experimental functional studies were performed [132]No clinical symptoms reported; sudden unexplained death [132]S173I518G > T [134]Missense, II (at the inter-dimer interface with III), Heterozygous [36]Disrupts CASQ2 filamentation by interfering with Ca²⁺-induced polymer formation at the inter-dimer interface, leading to defective Ca²⁺ buffering in the SR [36]CPVT-like and sudden unexplained death at young age [36]CPVT, Cardiac Arrest (CA)G112 + 5X339del16 [130]Deletion,II, III, and part of I,Homozygous [130]A 16 bp frameshift deletion in exon 3 introduces a premature stop codon, resulting in a truncated CASQ2 protein lacking ~ 260 downstream amino acids; this mutant cannot bind Ca²⁺ (owing to the loss of most of the acidic residues at the C-terminal), lacks key domains required for front-to-front and back-to-back interactions, and leads to decreased SR Ca²⁺ storage, reduced SR Ca²⁺ release, and increased spontaneous Ca²⁺ sparks [130]Exhibiting a severe form of CPVT, characterized by CA, exercise-induced syncope, rapid polymorphic ventricular tachycardia, and stress-triggered VT starting from young age [130]K206N618 A > C [135]Missense, II, Heterozygous,Heterozygous carrier [135]The mutation introduces an extra N-glycosylation site, leading to reduced Ca²⁺-binding capacity and impaired oligomer formation. This disrupts CASQ2 polymerization, enhances its interaction with RyR2, and increases the open probability of RyR2 Ca²⁺ channels. As a result, Ca²⁺ leak is elevated under both basal and stimulated conditions, SR Ca²⁺ concentration is reduced, and spontaneous Ca²⁺ release events in cardiomyocytes are more frequent. Despite these changes, CASQ2’s association with triadin remains unaffected [135]Syncope induced by emotion, stress, swimming and exercise since young age along with bradycardia and multiple extrasystoles despite normal QTc interval [135]W361X1083G > A [136]Nonsense,C-terminus,Homozygous [136]Causes premature truncation of the protein’s C-terminal domain, impairing its Ca²⁺-buffering function in the SR. This leads to excess free Ca²⁺, resulting in diastolic Ca²⁺ leak, which triggers arrhythmias under stress, manifesting as CPVT [136]Syncopal episodes since childhood, triggered by exercise and emotional stress. Initially misdiagnosed with epilepsy; ECG later showed left axis deviation and QT-U prolongation. Exercise testing induced polymorphic ventricular ectopy developing into polymorphic VT with syncope [136]CPVT, SDF189L567 C > G [137]Missense, II,Heterozygous [137, 138]Potential decrease in protein mobility [137–139]SD while exertion in the context of underlying, early-onset CPVTP308L923 C > T [131]Missense, III,Compound heterozygous [42, 131]Maintains dimerization yet fails to properly polymerize and interact with RyR2; causes conformational changes; Ca²⁺ selectivity is lost, and Mg2 + triggers aberrant aggregation [42, 125, 131, 140]Early-onset, exercise-induced syncope progressing to severe CPVT [131]CPVT, CA, SD-532 + 1G > A [118]Incorrect splicing,Homozygous intronic base pair change,While homozygous carrier individuals show symptoms, heterozygous siblings remain unaffected, indicating recessive inheritance [118, 128, 141]Leads to skipping exon 4, which introduces a frameshift and premature stop codon, preventing the production of full-length CASQ2 and resulting in a total loss of functional CASQ2 protein [118, 128, 141]Causes a severe form of CPVT in homozygous individuals, presenting in early childhood with syncope or CA triggered by exercise [118]K180R539 A > G [142]Missense, II,Heterozygous dominant inheritance [36, 142]The mutation maintains dimerization but disrupts inter-dimer interactions, impairing polymerization and resulting in a defective filament structure that alters interaction with IRE1α, while also impairing dynamic Ca²⁺ buffering in the SR without affecting total CASQ2 protein levels or overall SR Ca²⁺ content [36, 125, 142]It causes severe CPVT with early-onset symptoms. Clinical findings revealed exercise-induced ventricular arrhythmia and syncope [142]CA – Cardiac Arrest; Ca²⁺ – Calcium ion; CASQ2 – Calsequestrin 2; CICR – Calcium-Induced Calcium Release; CPVT – Catecholaminergic Polymorphic Ventricular Tachycardia; DADs – Delayed Afterdepolarizations; ECG – Electrocardiogram; IRE1α – Inositol-Requiring Enzyme 1 alpha; Mg²⁺ – Magnesium ion; QTc – Corrected QT Interval; RyR2 – Ryanodine Receptor 2; SD – Sudden Death; SR – Sarcoplasmic Reticulum; VT – Ventricular Tachycardia
Age and disease severity
The severity of CPVT exhibits marked age-dependent heterogeneity, with pediatric patients facing substantially higher risks of life-threatening arrhythmias during physical or emotional stress. The disease burden is particularly pronounced in children and adolescents, who demonstrate a 4-fold higher incidence of cardiac events compared to adults, with approximately one-third of patients experiencing their first symptomatic event before age 21 [143, 144]. These trends are primarily derived from studies of CPVT1, which represents the majority of reported cases.
In contrast, CPVT2 data remain limited, though available reports suggest a relatively later onset and milder arrhythmic phenotype in many patients. This apparent difference may reflect distinct molecular mechanisms, as RyR2 mutations directly alter the release channel, whereas CASQ2 mutations primarily affect luminal Ca²⁺ buffering.
Adult-onset CPVT cases, including those due to CASQ2 mutations, often show attenuated phenotypic expression with extended asymptomatic periods; however, they remain at risk of sudden cardiac death without warning. Notably, the 40-year arrhythmia-free survival rate for asymptomatic RyR2 mutation carriers is only 27%, underscoring the persistent risk across all age groups despite the apparent clinical quiescence in some adults [143], underscoring that age does not fully mitigate risk across CPVT subtypes. While extrapolation from CPVT1 to CPVT2 should be made with caution, both disorders share the central feature of stress-induced Ca²⁺ dysregulation as a driver of arrhythmic vulnerability.
Arrhythmias
Beyond CPVT, CASQ2 mutations are implicated in a broader spectrum of arrhythmias, including AF and sinus node dysfunction (SND) [74, 145, 146].
Normal cardiac automaticity depends on the cardiac sinoatrial node’s (SAN) pacemaker cells. Human SND is caused by cardiac pace-making disorder. SND is more common in older adults and is linked to irregular cardiac rhythms caused by compromised pace-maker function [147].
Sinoatrial node block, bradycardia/tachycardia syndrome, syncope, sinus bradycardia, and sinus arrest are among the symptoms that people with SND experience. Crucially, whether under stress or exercising, people with SND report experiencing chronotropic incompetence. SND can be inherited or linked to cardiovascular or systemic diseases. The only treatments available to patients with SND at this time are pace-maker implantation when necessary and the alleviation of arrhythmia symptoms [147].
CASQ2-deficient mouse models exhibit atrial ectopy, conduction block, and enhanced susceptibility to AF under adrenergic stimulation. As one study showed CPVT patients have both SAN and atrioventricular node dysfunction, and that these conditions are frequently linked to either spontaneous or exercise/catecholomine-induced AF. This suggests that the supraventricular tissues and the ventricle are involved in the disease’s background arrhythmogenic substrate as opposed to just the ventricular tissue as previously hypothesized. This could potentially be an indication for close follow-up with an implantable cardioverter-defibrillator (ICD) to monitor for AF [145]. Furthermore, structural remodeling, such as fibrosis in the sinoatrial node region, disrupts pacemaker activity and predisposes to SND [56].
Therapeutic approach
Efforts to restore CASQ2 functionality have led to diverse therapeutic strategies, including pharmacological interventions, molecular therapies, and emerging gene-based approaches aimed at improving Ca²⁺ homeostasis and reducing arrhythmogenic events.
Current interventions
Pharmacological
Reducing the frequency of arrhythmias is the primary objective of treatment. This can be accomplished by combining many strategies, including left cardiac sympathetic denervation, implantable cardiac defibrillators, medication treatments, and lifestyle modifications. Because lifestyle activities such as stressful circumstances, demanding workloads, and competitive sports lead to the release of catecholamines, which in turn can induce CPVT. As a result, guidelines suggest avoiding such activities.
Beta-blockers remain the first-line treatment for CASQ2-related arrhythmias [148]. These drugs mitigate adrenergic stimulation, which reduces intracellular Ca²⁺ overload and the risk of triggered arrhythmias. Propranolol and nadolol are widely prescribed [149, 150], but many patients experience breakthrough arrhythmias, necessitating adjunctive treatments. Flecainide, a sodium channel blocker, has shown efficacy in suppressing arrhythmias; however, its mechanisms of action remain unclear, with evidence pointing towards effects on both RyR2 and sodium channels, and to a minor extent on sodium-calcium exchanger, which collectively reduce DADs [151]. Despite their benefits, these pharmacological options often fail to provide comprehensive arrhythmia control [149, 152], highlighting the need for targeted therapies specifically addressing CASQ2 dysfunction.
Recent experimental evidence highlights melatonin as a promising adjunct in managing arrhythmias linked to impaired Ca²⁺ handling, particularly in heart failure settings. In acute ischemia-reperfusion (IR)-injured rabbit models, melatonin administration significantly reduced ventricular fibrillation maintenance and suppressed spatially discordant alternans - electrophysiological precursors of life-threatening arrhythmias [153, 154]. This antiarrhythmic effect was associated with enhanced SR Ca²⁺ handling, partly through upregulation of CASQ2 and SERCA2a expression, along with attenuation of phosphorylated phospholamban downregulation in failing hearts [153]. Notably, melatonin’s beneficial effects on Ca²⁺ homeostasis were partially counteracted by therapeutic hypothermia, underscoring the importance of temperature-sensitive mechanisms in its efficacy.
However, these findings were derived from heart-failure and ischemic models rather than CASQ2-mutant systems. In CPVT2, most mutations are homozygous and result in structurally defective CASQ2 proteins, meaning that simply increasing the amount of mutated protein may not restore normal RyR2 interactions or Ca^2+^ buffering. Therefore, the direct benefit of melatonin-induced CASQ2 upregulation in CPVT2 remains uncertain and should be interpreted cautiously.
While not traditionally classified as an antiarrhythmic drug, melatonin’s ability to improve SR Ca²⁺ storage capacity via CASQ2 modulation in experimental settings suggests potential utility in conditions marked by defective Ca²⁺ buffering and diastolic Ca²⁺ leak. In CPVT2, however, greater SR Ca²⁺ loading through SERCA2a upregulation could theoretically worsen RyR2 hyperactivity, so further research is needed to understand this effect. Its antioxidant and anti-inflammatory properties further support cardioprotection in IR injury models, positioning melatonin as a multifunctional agent that may complement existing pharmacotherapies for arrhythmias related to impaired Ca²⁺ handling, although its role in CASQ2-related CPVT2 remains speculative at present [155].
Non-pharmacological
Patients with CPVT require comprehensive lifestyle and medical management to mitigate arrhythmia risks. Current guidelines strongly advise against competitive sports and high-intensity physical activities such as sprinting or basketball due to the well-documented risk of adrenergically-triggered ventricular arrhythmias. However, carefully supervised mild-to-moderate aerobic exercise like walking or light cycling may be permissible for select patients who demonstrate excellent arrhythmia control on medication and have no history of cardiac events [156]. Regular follow-up with a cardiologist specializing in inherited arrhythmias is crucial, particularly during high-risk periods like puberty, pregnancy, and growth spurts when hormonal changes may increase arrhythmia susceptibility.
For patients with refractory arrhythmias or multiple ICD shocks despite optimal pharmacotherapy, left cardiac sympathetic denervation (LCSD) represents an important intervention [157]. This surgical procedure involves the resection of the lower half of the left stellate ganglion and the first thoracic ganglia (T1-T4), significantly reducing norepinephrine release to the heart. While the majority of clinical evidence comes from patients with CPVT1, LCSD is also considered for high-risk CPVT2 patients, effectively reducing the burden of life-threatening arrhythmias by blunting the sympathetic trigger [158].
Genetic counseling forms an essential component of CPVT care, providing family risk assessment through cascade genetic testing and discussing reproductive options including prenatal diagnosis and preimplantation genetic testing. Comprehensive diagnostic evaluation combines exercise stress testing, which provokes diagnostic ventricular arrhythmias in approximately 75% of cases, with molecular genetic testing that identifies pathogenic variants in 60–70% of clinically diagnosed patients [106]. Emerging therapeutic options including flecainide for RYR2-mediated CPVT and novel Rycal compounds show promise in clinical trials, though beta-blockers remain the mainstay of therapy. All patients require individualized management plans incorporating these elements to optimize outcomes while maintaining quality of life [106, 156].
Emerging therapies and future directions
CASQ2 stabilizers
Emerging pharmacological treatments for CPVT aim to correct the pathways responsible for arrhythmogenesis. Research has highlighted the effectiveness of K201, a 1,4-benzothiazepine derivative, in stabilizing calstabin2-RyR2 binding, likely due to its structural similarity to diltiazem [159, 160]. This stabilization has been shown to prevent arrhythmias.
Another innovative agent, ent-(+)-verticilide, a synthetic derivative of the insecticide verticilide, was found to reduce delayed after-depolarizations by selectively inhibiting RyR2 Ca²⁺ release in murine cardiac cells with CASQ2 mutations. While its exact mechanism of action remains unclear, it appears to act independently of RyR2 phosphorylation or interactions with accessory proteins [161]. Conversely, both dantrolene and K201 target two of the three known mechanisms of RyR2 dysfunction. Dantrolene has shown success in reducing arrhythmias in patients with RyR2 domain defects, while clinical trials for K201 and ent-(+)-verticilide have yet to be conducted [162].
Gene therapy and stem cell models
Gene therapy has emerged as a revolutionary approach for addressing CASQ2-related pathologies, with adeno-associated virus (AAV) vectors becoming the leading delivery platform due to their cardiac tropism and long-term transgene expression. Preclinical studies using AAV9-CASQ2 in CPVT mouse models demonstrated > 80% restoration of CASQ2 protein levels, resulting in complete suppression of ventricular arrhythmias during adrenergic challenge and improved 6-month survival rates from 40% to 90% [163–169]. Notably, AAV-mediated CASQ2 expression was shown to functionally compensate for both loss-of-function mutations and CASQ2 knockout phenotypes by normalizing SR Ca^2+^ buffering capacity [163–169].
Complementary to gene therapy, human-induced pluripotent stem cell (hiPSC) models from CPVT patients have enabled unprecedented mechanistic studies of CASQ2 dysfunction. These patient-specific cardiomyocyte models recapitulate key disease features including abnormal Ca^2+^ transients and delayed afterdepolarizations, serving as platforms for drug screening [170]. Recent hiPSC studies identified flecainide and carvedilol as particularly effective in CASQ2-deficient cells, with combination therapy showing synergistic benefits [171, 172].
Recent advances in iPSC modeling have enabled precise investigation of CASQ2-related pathophysiology. A new isogenic Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas9-corrected iPSC line derived from a CPVT patient carrying a heterozygous mutation in CASQ2 has been established, offering a powerful platform to dissect molecular mechanisms and evaluate therapeutic interventions [173, 174]. These gene-edited iPSCs were shown to retain normal karyotype, pluripotency markers, and absence of off-target mutations, making them suitable for differentiating into cardiomyocytes that recapitulate patient-specific Ca²⁺ handling abnormalities [175]. Such models allow for high-fidelity comparisons between mutant and corrected lines, supporting the development of precision therapies targeting CASQ2 dysfunction and associated arrhythmias.
Parallel therapeutic development includes small molecule modulators targeting the CASQ2-RyR2 interaction, such as the Rycal compound S107 which stabilizes the RyR2-calstabin2 complex and reduces diastolic Ca^2+^ leak in CASQ2-mutant cardiomyocytes [176, 177]. Second-generation derivatives with improved bioavailability are currently in Phase I/II trials for CPVT. These approaches collectively address the fundamental pathophysiology of CASQ2 deficiency: impaired Ca^2+^ buffering, aberrant RyR2 regulation, and consequent pro-arrhythmic Ca^2+^ waves.
Challenges in developing CASQ2-Targeted therapies
The development of CASQ2-targeted therapies faces several challenges. Genetic heterogeneity among CASQ2 mutations complicates the creation of universal treatments, requiring personalized approaches [178, 179]. Delivering therapies specifically to cardiac tissue without affecting skeletal muscle isoforms presents additional obstacles [163, 180]. Moreover, the long-term safety and efficacy of emerging therapies, including gene-based interventions, remain critical areas of concern. Addressing these challenges will be essential for translating preclinical successes into viable clinical treatments [114].
Therapeutic approaches targeting CASQ2 have advanced significantly, encompassing pharmacological, molecular, and gene-based strategies. While current interventions offer partial symptomatic relief and reduced risk of adverse events, emerging therapies hold the promise of addressing the underlying molecular dysfunctions of CASQ2. Overcoming challenges related to genetic variability, delivery specificity, and long-term safety will be pivotal in realizing the full potential of these innovative treatments and improving outcomes for patients with CASQ2-related arrhythmias [114, 163, 180].
These strategies are summarized in Table 2, which outlines current, emerging, and experimental approaches for targeting CASQ2-related dysfunction.
Table 2. Therapeutic strategies for CASQ2-Related catecholaminergic polymorphic ventricular tachycardia (CPVT)CategoryTherapeutic StrategyMechanism of ActionKey InsightsCurrent InterventionsBeta-blockers (e.g.,** propranolol**,** nadolol)**Block β-adrenergic stimulation to reduce intracellular Ca²⁺ overloadFirst-line treatment for CPVT; reduce risk of triggered arrhythmias but may not fully suppress symptoms in all patients Flecainide Blocks sodium channels and inhibits spontaneous Ca²⁺ release through RyR2Reduces delayed afterdepolarizations (DADs); often used in combination with beta-blockers for better control Lifestyle Modifications Avoid physical/emotional stress that triggers adrenergic surgesCritical for preventing arrhythmic episodes; high-intensity sports discouraged, light activity allowed under supervision Implantable Cardioverter-Defibrillator (ICD) Provides immediate correction of life-threatening arrhythmiasConsidered for high-risk or drug-refractory cases Left Cardiac Sympathetic Denervation (LCSD) Surgical resection of left stellate/T1-T4 ganglia to reduce cardiac norepinephrine releaseReserved for high-risk, drug-refractory patients; reduces arrhythmia burden and ICD shocks by blunting sympathetic trigger. Genetic Counseling & Testing Identifies carriers and guides reproductive planningEnables early diagnosis in family members; supports personalized care strategiesEmerging Therapies K201 (JTV519) Enhances RyR2-calstabin2 binding, stabilizing RyR2 channelPrevents pathological Ca²⁺ leak; promising in preclinical models; related structurally to diltiazem Dantrolene Restores RyR2 interdomain stability and normalizes gatingShown to reduce ventricular arrhythmias in mutant RyR2 models; targets defective channel conformation ent-(+)-Verticilide Selectively inhibits RyR2-mediated Ca²⁺ releaseReduces DADs in CASQ2-mutant cells; mechanism independent of RyR2 phosphorylation or accessory proteins S107 (Rycal compound) Stabilizes RyR2–calstabin2 complex to reduce Ca²⁺ leakReduces diastolic Ca²⁺ leak in CASQ2-deficient cardiomyocytes; second-generation derivatives in early clinical trials Melatonin Enhances SR Ca²⁺ handling through upregulation of CASQ2 and SERCA2a; modulates phospholamban phosphorylationDemonstrated to reduce ventricular fibrillation and Ca²⁺ alternans in IR injury models; also offers antioxidant and anti-inflammatory cardioprotection. Evidence derived from heart-failure models; potential relevance to CASQ2-related CPVT2 remains speculative.Gene Therapy AAV9-CASQ2 Gene Transfer Replaces defective CASQ2 gene using cardiac-targeted AAV9 vectorDemonstrated > 80% restoration of CASQ2 expression and suppression of arrhythmias in CPVT mouse models; improved survival ratesPatient-Specific Models hiPSC-derived Cardiomyocytes Replicate patient-specific CASQ2 defects for drug screeningReveal abnormal Ca²⁺ handling and arrhythmogenic behavior; enable evaluation of personalized drug responses (e.g., flecainide + carvedilol synergy) CRISPR/Cas9-Corrected iPSC Model Isogenic mutation correction in patient-derived iPSCsProvides high-fidelity platform for comparing mutant vs. corrected lines; supports discovery of targeted therapies for CASQ2-related arrhythmiasChallenges Genetic Heterogeneity Wide range of CASQ2 mutations leads to variable functional impactLimits the development of universal therapies; personalized strategies are needed Tissue-Specific Targeting Avoiding off-target effects in non-cardiac tissues (e.g., skeletal muscle)Critical for gene therapies and small molecules with systemic delivery Long-Term Safety & Efficacy Especially relevant for gene-based and novel molecular interventionsRequires careful long-term follow-up and monitoring of therapeutic durability and side effectsAAV – Adeno-associated virus; AAV9 – Adeno-associated virus serotype 9; β – Beta; Ca²⁺ – Calcium ion; CASQ2 – Calsequestrin 2; CPVT – Catecholaminergic Polymorphic Ventricular Tachycardia; DADs – Delayed Afterdepolarizations; ICD – Implantable Cardioverter-Defibrillator; IR – Ischemia-Reperfusion; LCSD – Left Cardiac Sympathetic Denervation; RyR2 – Ryanodine Receptor 2; SERCA2a – Sarcoplasmic Reticulum Ca²⁺-ATPase 2a; SR – Sarcoplasmic Reticulum; hiPSC – human-induced Pluripotent Stem Cell; CRISPR – Clustered Regularly Interspaced Short Palindromic Repeats; iPSC – induced Pluripotent Stem Cell; CASQ – Calsequestrin; VT – Ventricular Tachycardia
Conclusion
Calsequestrin 2 is a key protein in cardiac muscle physiology, serving as the primary Ca²⁺-binding protein in the SR. Its role extends beyond Ca²⁺ buffering, as it actively regulates Ca²⁺ release through interactions with RyRs, triadin, and junctin. These interactions ensure precise Ca²⁺ handling, which is fundamental for maintaining cardiac rhythm and contraction efficiency. The structural organization of CASQ2, including its thioredoxin-like domains and intrinsically disordered C-terminal region, allows it to dynamically respond to fluctuations in luminal Ca²⁺ concentrations, ensuring proper EC coupling.
Dysfunction in CASQ2 leads to severe cardiac pathologies, most notably CPVT, an inherited arrhythmogenic disorder that significantly increases the risk of SCD. Mutations in the CASQ2 gene disrupt its Ca²⁺-binding capacity and polymerization dynamics, leading to spontaneous Ca²⁺ release, delayed afterdepolarizations, and ventricular arrhythmias. Despite advances in pharmacological treatments such as beta-blockers and flecainide, many patients remain at high risk, highlighting the urgent need for more targeted therapies.
Emerging therapeutic strategies, including CASQ2 stabilizers, gene therapy, and small-molecule modulators, offer promising avenues for restoring normal Ca²⁺ homeostasis. While challenges remain, such as genetic heterogeneity and tissue-specific targeting, ongoing research is paving the way for precision medicine approaches in CASQ2-related arrhythmias. Future advancements in molecular therapies and gene-editing techniques may ultimately provide curative treatments, transforming the management of CASQ2-associated cardiac diseases. Through continued exploration, CASQ2 remains not only a fundamental component of Ca²⁺ regulation but also a key target in the quest to mitigate arrhythmogenic cardiac disorders.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Bers DM, Eisner DA, Valdivia HH (2003) Sarcoplasmic Reticulum Ca 2 Heart Fail Roles Diastolic Leak Ca 2 Transp 10.1161/01.RES.0000091871.54907.6B 14500331 · doi ↗ · pubmed ↗
- 2Carlo Napolitano AM, Bloise R, Silvia G, Priori (2022) Catecholaminergic polymorphic ventricular tachycardia. University of Washington, Seattle 20301466 · pubmed ↗
- 3Zhang J-c et al (2018) Calcium-Mediated Oscillation in membrane potentials and atrial-Triggered activity in atrial cells of Casq 2R 33Q/R 33Q mutation mice. Frontiers in Physiology, pp 9–201810.3389/fphys.2018.01447 PMC 622435930450052 · doi ↗ · pubmed ↗
- 4Fujisawa T et al (2019) A homozygous CASQ 2 mutation in a Japanese patient with catecholaminergic polymorphic ventricular tachycardia, vol 2019. Case Reports in Genetics, p 9056596. 110.1155/2019/9056596 PMC 634126730729048 · doi ↗ · pubmed ↗
- 5Priori SG et al (2015) ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The task force for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur Heart J, 2015. 36(41): pp. 2793–286710.1093/eurheartj/ehv 31626320108 · doi ↗ · pubmed ↗