Advances in chimeric antigen receptor-natural killer cell therapy: from mechanisms and preclinical studies to clinical application
Tianyuan Ren, Fengjiang Wang, Xuan Liu, Jun Guo, Sitan Xie

TL;DR
This review explores CAR-NK cell therapy as a promising alternative to CAR-T for cancer treatment, highlighting recent advances and challenges in its development and application.
Contribution
The paper provides a comprehensive synthesis of CAR-NK engineering strategies and clinical progress, distinguishing it from CAR-T approaches.
Findings
CAR-NK cells offer advantages like lower risk of cytokine release syndrome and potential for off-the-shelf production.
Strategies like Fc-binding domains and cytokine armoring are being explored to enhance CAR-NK efficacy in tumor microenvironments.
Challenges include in vitro expansion limitations and immunosuppressive tumor environments, with potential technical solutions discussed.
Abstract
Chimeric antigen receptor T-cell therapy (CAR-T) has revolutionized cancer treatment, yet its application remains limited by high costs, safety concerns, and challenges in solid tumors. Natural killer (NK) cells offer a promising alternative due to their innate tumor-killing capacity, diverse cell sources, lower risk of graft-versus-host disease and cytokine release syndrome, and potential for “off-the-shelf” production. This review synthesizes recent advances in CAR-NK, focusing on NK-specific CAR engineering strategies, preclinical models across hematological and solid malignancies, and the latest clinical trials up to 2025. We highlight distinctive CAR-NK optimization approaches, such as integration of Fc-binding domains, cytokine armoring, and strategies to overcome tumor microenvironment mediated resistance, that distinguish CAR-NK from CAR-T platforms. Key challenges, including…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
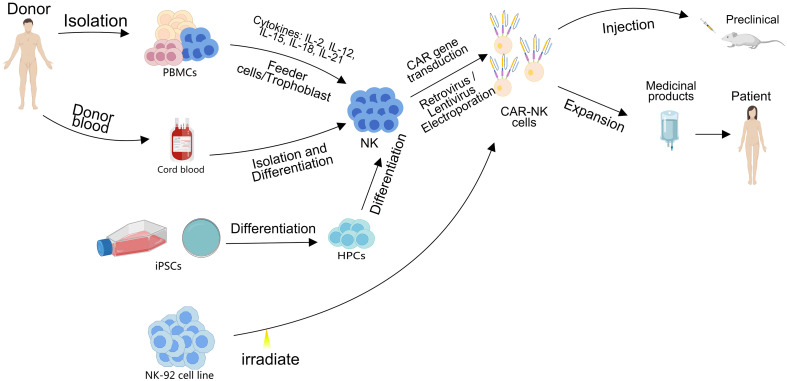 Figure 1
Figure 1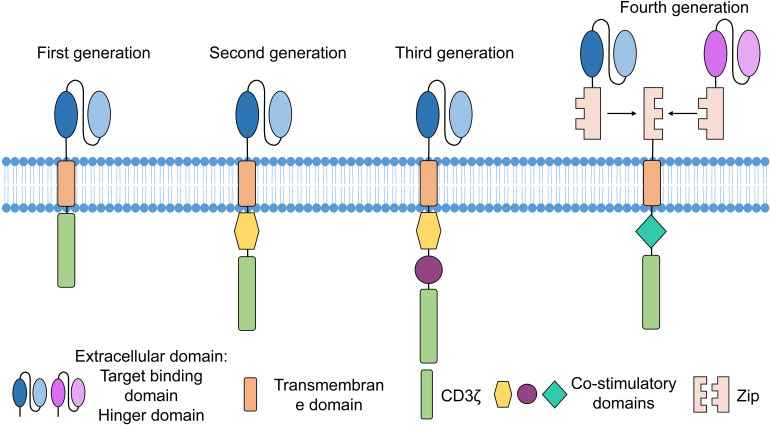 Figure 2
Figure 2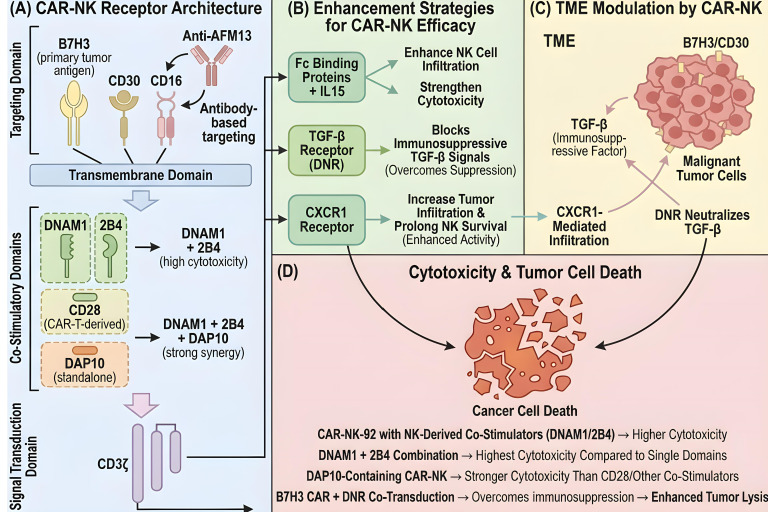 Figure 3
Figure 3| Name | Target | Manufacturer | Disease | Country | References |
|---|---|---|---|---|---|
| Kymriah | CD19 | Novartis | B-ALL/B-NHL | USA | ( |
| Yescarta | CD19 | Kite Pharma | B-NHL | USA | ( |
| Tecartus | CD19 | Kite Pharma | R/R MCL/B-ALL | USA | ( |
| Breyanzi | CD19 | Bristol Myers Squibb | LBCL | USA | ( |
| Abecma | BCMA | Bristol Myers Squibb | R/R MM | USA | ( |
| Carteyva | CD19 | JW Therapeutics | R/R LBCL | China | ( |
| Carvykti | BCMA | LEGEND Biotech | R/R MM | USA | ( |
| Fucaso | BCMA | IASO Biotherapeutics | R/R MM | China | ( |
| NexCAR19™ | CD19 | ImmunoACT | R/R B-cell malignancies/B-ALL | India | ( |
| Zevor-cel | BCMA | Carsgen Therapeutics | R/R MM | China | ( |
| Yuanduida | CD19 | Juventas Cell Therapy Ltd | R/R B-ALL | China | ( |
| Aucatzyl | CD19 | University College London/Autolus | R/R B-ALL | USA/Europe | ( |
| HICARA | CD19 | Shanghai Hengrundasheng Biotechnology Co., Ltd. | R/R LBCL | China | ( |
| Malignancy type | Target antigen | CAR design/Engineering strategy (Conceptual advance) | NK cell source | Key preclinical findings |
|---|---|---|---|---|
| Hematological Malignancies | ||||
| MM | BCMA, GPRC5D | Dual-Targeting CAR | Not Specified | Superior cytotoxicity and reduced tumor recurrence compared to single-target CAR-NK cells. |
| AML | CD33 | Checkpoint Knockout (NKG2A via CRISPR/Cas9) | Not Specified | Enhanced |
| AML | CD123 | Standard CAR | Not Specified | Induced lower acute toxicity with anti-leukemic efficacy comparable to CAR-T cells. |
| AML/Broad Hematological | NKG2D | Cytokine Armoring (mbIL-15) | Not Specified (Nkarta’s NKX101) | Robust and sustained cytolytic activity independent of exogenous cytokine support. |
| AML | CD133/CD16 | Bifunctional CAR (Engages endogenous ADCC) | Not Specified | ~80% cytotoxicity against AML cells, superior to single-target CD33-CAR-NK cells. |
| Solid Tumors | ||||
| HCC | GPC3 | TME Resistance (sPD-L1 blockade) | NK-92 | Potent cytotoxicity; co-administration of sPD-L1 variant reversed inhibition and enhanced efficacy. |
| GPC3 | NK-specific co-stimulation (DNAM1 + 2B4) | Not Specified | Further elevated cytotoxicity of GPC3-CAR-NK cells. | |
| CD147, GPC3 | Bispecific CAR (Dual antigen recognition for safety) | Not Specified | Effective tumor elimination; no toxicity in CD147 transgenic models, suggesting safety for off-the-shelf use. | |
| c-MET | Target Validation | Primary NK | Superior cytotoxicity against high c-MET-expressing HCC cells. | |
| OC | MSLN | Standard Target Validation | NK-92 | Potent and specific anti-tumor activity |
| MSLN | Cytokine Signaling Enhancement (Neo-2/15 agonist) | Not Specified | Superior proliferation, cytolytic capacity, and | |
| GPC3 | Off-the-shelf Platform (iPSC-derived) | iPSC-NK/ILC | Significant survival benefit in models with no acute toxicity or tumorigenicity. | |
| CD24 | Optimized scFv (“SW11”) in 3rd gen CAR | NK-92 | Potent, specific activity against OC cell lines and primary patient cells. | |
| CLDN6 | Standard Target Validation | NK-92MI | Robust anti-tumor effects against CLDN6+ OC cells. | |
| GC | MSLN | Standard CAR | NK-92 | Effective tumor lysis and extended survival in PDX models. |
| HER2 | Combination Therapy (with apatinib) | NK-92 | Efficacy in small tumors; apatinib improved infiltration. Highlights limitation in large, established tumors. | |
| PD1/NKG2D | Novel Receptor Design (DTCR: PD1-DAP10/NKG2D) | NK-92 | Dramatically augmented cytotoxicity and therapeutic enhancement | |
| HER2 | Enhanced Innate Immunity (High-affinity, cleavage-resistant CD16) | Placental HSC-NK (CYNK-101) | Next-generation off-the-shelf candidate with enhanced proliferative potential. | |
| GBM | Multiple/CD73 | TME Modulation (Local secretion of anti-CD73 scFv) | Not Specified | Overcame TME suppression and attenuated antigen escape via local adenosine pathway inhibition. |
| GFR/EGFRvIII | Dual-Targeting CAR | Not Specified | Significant tumor shrinkage and extended survival after intracranial administration. | |
| EGFRvIII | Improved Trafficking (CXCR4 overexpression) | Not Specified | Enhanced tumor accumulation and survival benefits. | |
| HER2 | Standard Target Validation | Not Specified | Effective cytolysis and robust tumor growth inhibition in models. | |
| EGFR | Combination Therapy (with oncolytic virus OV-IL15C) | Off-the-shelf CAR-NK | Synergistic effect, increased immune cell infiltration and persistence. | |
| DIPG | GD2 | Standard Target Validation | NK-92 | Effective killing of high GD2-expressing DIPG cells; target-dependent efficacy. |
| Prostate Cancer | PSMA | Combination Therapy (with anti-PD-L1) | NK-92 | Enhanced antitumor efficacy in castration-resistant models. |
| TNBC | CD44v6 | Standard Target Validation | NK cells | Targeted therapy for triple-negative breast cancer. |
| NK source | Target | CAR structure | Cancer | Phase | Status | NCT No. | Study start | Country |
|---|---|---|---|---|---|---|---|---|
| Unknown | Unknown | Unknown | Hematopoietic/Lymphoma | III | Completed | Nov. 2008 | USA | |
| Unknown | CD30 | Unknown | Lymphomas | I/II | Unknown status | Mar. 2014 | China | |
| NK-92 | CD7 | scFv-CD28-4-1BB-CD3ζ | T cell malignancies/AML | I/II | Unknown | Mar. 2016 | China | |
| NK-92 | CD19 | scFv-CD28-4-1BB-CD3ζ | Leukemia and Lymphoma | I/II | Recruiting | Sep. 2016 | China | |
| NK-92 | CD33 | scFv-CD28-4-1BB-CD3ζ | AML | I/II | Unknown | Oct. 2016 | China | |
| Cord blood | CD19 | scFv-CD28-CD3ζ-iCasp9-IL15 | B-cell lymphoma | I/II | Completed | Jun. 2017 | USA | |
| Unknown | CD19/CD22 | Unknown | Refractory B-cell lymphoma | Early Phase I | Unknown | Feb. 2019 | Unknown | |
| Unknown | CD19 | Unknown | Refractory B-cell lymphoma | Early Phase I | Unknown | Mar. 2019 | Unknown | |
| Unknown | CD22 | Unknown | Refractory B-cell lymphoma | Early Phase I | Unknown | Mar. 2019 | Unknown | |
| NK-92 | BCMA | scFv-CD8αTM-4-1BB-CD3ζ | MM | I/II | Unknown | May. 2019 | China | |
| Unknown | CD7 | Unknown | Leukemia and lymphoma | I | Unknown | Aug. 2019 | China | |
| Cord blood | CD19 | scFv-CD28-CD3ζ-iCasp9-IL15 | B-cell lymphoma | I/II | Withdrawn | Oct. 2019 | USA | |
| PB-NK | NKG2D-L | NKG2D-OX40-CD3ζ-mblL-15 | R/R AML/MDS | I | Recruiting | Sep. 2020 | USA | |
| Unknown | CD7 | Unknown | Hematological malignancies | I | Completed | Sep. 2020 | China | |
| Unknown | CD33/CLL1 | Unknown | AML | Early Phase I | Recruiting | Nov. 2020 | China | |
| Unknown | CD7 | Unknown | T-Lymphoblastic leukemia | I | Unknown | Nov. 2020 | China | |
| Unknown | CD19 | Unknown | NHL | Early Phase I | Not yet recruiting | Dec. 2020 | China | |
| Unknown | CD19 | Unknown | B-cell lymphoma | I | Recruiting | Mar. 2021 | China | |
| Cord blood | CD19 | Unknown | Lymphoblastic leukemia/NHL | I | Recruiting | Apr. 2021 | China | |
| PB-NK | CD19 | Unknown | NHL | I | Recruiting | May. 2021 | China | |
| PB-NK | CD19 | CD19 scFv-CD8aTM-OX40-CD3ζ-T2A-IL15 | B-cell malignancies | I | Recruiting | Aug. 2021 | USA | |
| Cord blood | BCMA | Unknown | MM | Early Phase I | Recruiting | Oct. 2021 | China | |
| Cord blood | NKG2D-L | Unknown | AML | NA | Terminated | Oct. 2021 | China | |
| iPSC (FT576) | BCMA | scFv-NKG2D-2B4-CD3ζ-IL15/R-hnCD16 | MM | I | Recruiting | Nov. 2021 | USA | |
| Unknown | Unknown | Unknown | B-cell leukemia/B-cell lymphoma | I/II | Unknown | Nov. 2021 | China | |
| Unknown | Unknown | Unknown | B-cell acute leukemia/B-cell lymphoma | I | Recruiting | Nov. 2021 | China | |
| PB-NK | CD19 | Unknown | Lymphocytic leukemia/NHL | I | Recruiting | May. 2022 | China | |
| Unknown | CD19 | Unknown | ALL | I | Completed | Jul. 2022 | China | |
| Cord blood | CD19 | Unknown | NHL | I | Recruiting | Sep. 2022 | China | |
| Unknown | CD123 | Unknown | R/R AML | Early Phase I | Recruiting | Oct. 2022 | China | |
| Unknown | CD19 | Unknown | Relapsed NHL/B-cell lymphoma/B-cell leukemia/DLBCL | I | Recruiting | Oct. 2022 | USA | |
| Unknown | CD19 | Unknown | B-cell lymphoma/B-cell leukemia | I/II | Withdrawn | Oct. 2022 | China | |
| Unknown | Unknown | Unknown | AML | I | Recruiting | Oct. 2022 | China | |
| Cord blood | CAR.70 | Unknown | B cell lymphoma/MDS/AML | I/II | Recruiting | Nov. 2022 | USA | |
| Unknown | BCMA | Unknown | MM | Early Phase I | Recruiting | Nov. 2022 | China | |
| Unknown | CD19/CD70 | Unknown | NHL | I | Recruiting | Dec. 2022 | China | |
| Unknown | CD19 | Unknown | Refractory B-cell hematologic malignancies | I | Recruiting | Dec. 2022 | China | |
| Unknown | CD19 | Unknown | B-cell lymphocytic leukemia/lymphoma | I/II | Recruiting | Dec. 2022 | China | |
| Unknown | CD33 | Unknown | AML | I | Recruiting | Dec. 2022 | China | |
| PB-NK | CD19, CD79 and CD123 | Unknown | Myelo or lymphoproliferative disorders | Not applicable | Recruiting | Dec. 2022 | European | |
| Cord blood | CD19/CD70 | Unknown | R/R NHL | I/II | Recruiting | Jan. 2023 | China | |
| Unknown | CD19 | Unknown | DLBCL | Early Phase I | Recruiting | Mar. 2023 | China | |
| Unknown | CD19 | Unknown | Leukemia/B-cell lymphoma | Early Phase I | Recruiting | Mar. 2023 | China | |
| Unknown | NKG2D | Unknown | AML | NA | Recruiting | Mar. 2023 | China | |
| Unknown | BCMA | Unknown | MM | Early Phase I | Recruiting | Jul. 2023 | China | |
| Unknown | CD33/CLL1 | Unknown | AML | I | Not yet recruiting | Aug. 2023 | China | |
| Unknown | CD19 | Unknown | SLE | Early Phase I | Recruiting | Aug. 2023 | China | |
| Unknown | CD123 | Unknown | BPDCN | I/II | Recruiting | Aug. 2023 | China | |
| Unknown | CLL1 | Unknown | AML | I | Recruiting | Sep. 2023 | China | |
| CB-NK | CD5 | Unknown | Hematological malignancy | I/II | Recruiting | Nov. 2023 | USA | |
| Unknown | CD19 | Unknown | R/R NHL | I | Recruiting | Nov. 2023 | USA | |
| Unknown | CD123 | Unknown | R/R AML | Early Phase I | Recruiting | Dec. 2023 | China | |
| Unknown | CD19 | Unknown | Resistant B-cell ALL | I/II | Not yet recruiting | Aug. 2024 | China | |
| Unknown | CD19/ | Unknown | SLE | Early Phase I | Recruiting | Jan. 2025 | China | |
| iPSC | CD19 | Unknown | Refractory B cell-mediated autoimmune disease | I | Recruiting | Feb. 2025 | USA | |
| iPSC | CD19 | Unknown | B-Cell Malignancies | I | Active, not recruiting | Jan. 2023 | USA |
| NK source | Target | CAR structure | Cancer | Phase | Status | No. NCT | Study start | Country |
|---|---|---|---|---|---|---|---|---|
| NK-92 | MUC1 | scFv-CD28-4-1BB-CD3ζ | MUC1positive solid tumor | I/II | Unknown | Jul. 2016 | China | |
| PB-NK | NKG2D-L | scFv-CD8αTM-CD3ζ | Solid Tumor | I | Unknown | Jan. 2018 | China | |
| Unknown | PSMA | PSMA CAR-NK | Metastatic castration-resistant prostate cancer | EARLY I | Recruiting | Dec. 2018 | China | |
| Unknown | Unknown | Unknown | Cancer | I/II | Recruiting | Jan. 2019 | China | |
| PB-NK | Mesothelin | Unknown | Epithelial OC | EARLY I | Unknown | Mar. 2019 | Unknown | |
| Primary NK cells | ROBO1 | scFv-CD8αTM-4-1BB-CD3ζ | Solid tumor | I/II | Unknown | May. 2019 | China | |
| NK-92 | ROBO1 | scFv-CD8αTM-4-1BB-CD3ζ | Pancreatic cancer | I/II | Unknown | May. 2019 | China | |
| NK-92 | ROBO2 | scFv-CD8αTM-4-1BB-CD3ζ | Solid tumor | I/II | Unknown | May. 2019 | China | |
| Unknown | NKG2D,ACE2 | Unknown | COVID-19 | I/II | Unknown | Feb. 2020 | China | |
| NK-92 | PD-L1 | Anti-PD-L1 CAR-IL-2-CD16 (PD-L1 t-haNK) | Pancreatic cancer | II | Recruiting | Jul. 2020 | Unknown | |
| raNK (ready-to-use allogeneic NK) | 5T4 | Unknown | Locally advanced/Metastatic solid tumors | EARLY I | Recruiting | Nov. 2021 | China | |
| Unknown | 5T4 | Unknown | Advanced solid tumors | EARLY I | Recruiting | Dec. 2021 | China | |
| NK-92 | PD-L1 | Anti-PD-L1 CAR-IL-2-CD16 (PD-L1 t-haNK) | GEJ Cancer/Advanced head and neck cancer | II | Recruiting | Dec. 2021 | USA | |
| Unknown | NKG2D-L | Unknown | Refractory mastatic colorectal cancer | I | Recruiting | Dec. 2021 | China | |
| PB-NK | Claudin6 | CLDN6-CAR-NK | Stage IV OC/Refractory testicular cancer | I/II | Recruiting | Jun. 2022 | China | |
| NK-92 | DLL3 | Unknown | SCLC | I | Recruiting | Sep. 2022 | China | |
| Unknown | SZ011 | Unknown | Advanced triple negative breast cancer | EARLY I | Not Yet Recruiting | Feb. 2023 | China | |
| CB-NK | CAR.70 | Unknown | Advanced renal cell carcinoma, mesothelioma, osteosarcoma | I/II | Recruiting | Mar. 2023 | USA | |
| Unknown | NKG2D | Unknown | OC | NA | Recruiting | Mar. 2023 | China | |
| Unknown | SZ003 | Unknown | Advanced hepatocellular carcinoma | NA | Not Yet Recruiting | May. 2023 | Unknown | |
| Unknown | SZ011 | Unknown | Epithelial OC | EARLY I | Not Yet Recruiting | Jun. 2023 | Unknown | |
| CB-NK | TROP2 | Unknown | Pancreatic cancer/OC/Adenocarcinoma | I/II | Not Yet Recruiting | Oct. 2023 | USA | |
| CB-NK | TROP2 | Unknown | Solid tumor | I | Not Yet Recruiting | Oct. 2023 | USA | |
| CB-NK | TROP2 | TROP2 CAR engineered IL-15- transduced CB-NK | Colorectal cancer | I | Recruiting | Dec. 2024 | USA | |
| Unknown | PSMA | Unknown | Metastatic castration-resistant prostate cancer | Early Phase I | Not yet recruiting | Aug. 2025 | China | |
| Unknown | CD70 | Unknown | Refractory clear cell renal cell carcinoma | I | Recruiting | Sep. 2025 | USA |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCAR-T cell therapy research · Immune Cell Function and Interaction · Immunotherapy and Immune Responses
Introduction
1
Current status and limitations of CAR-T
1.1
With the growing number of chimeric antigen receptor T-cell (CAR-T) products entering the market, adoptive immune cell therapy has garnered substantial attention. As of September 2025, 14 CAR-T products have secured global marketing approval (Table 1) (1–10), 7 of which are approved for use in China, a milestone that solidifies the country’s pivotal role in the global CAR-T landscape.
Despite these advances, autologous CAR-T is hampered by inherent limitations. These include elaborate, cost-prohibitive manufacturing processes with prolonged turnaround times, and a substantial risk of graft-versus-host disease (GvHD) (11). Additionally, CAR-T is prone to exhaustion (12), which can trigger severe cytokine release syndrome (CRS) (13) and neurotoxicity (14). Their therapeutic efficacy against solid tumors also remains suboptimal, primarily due to the immunosuppressive nature of the tumor microenvironment (TME) and the poor infiltration capacity of CAR-T into tumor tissues. These unmet clinical needs have spurred intensive exploration of alternative adoptive immune cell-based therapeutic strategies.
Mechanisms and advantages of CAR-NK
1.2
Since Kuwana (1987) (15) and Gross (1989) (16) first proposed the concept of chimeric antigen receptors (CARs), CAR-based cellular therapies have expanded across multiple disease fields, from hematological and solid tumors to autoimmune disorders (17). The clinical success of CAR-T has further accelerated the development of CAR-natural killer (CAR-NK) cell technology.
NK cells inherently eliminate tumor cells independent of prior antigen priming, thus CAR-NK is widely regarded as a highly effective strategy for tumor immunotherapy (18). While CAR structure design for NK cells shares similarities with CAR-T, both involve genetic element delivery and ex vivo expansion of engineered cells (19), and the unique biological characteristics of NK cells endow CAR-NK with distinct advantages over CAR-T.
Multiple sources are exploited for generating allogeneic CAR-NK, including peripheral blood (PB) from healthy donors (20, 21), umbilical cord blood (UCB) (22), induced pluripotent stem cells (iPSCs) (21, 23, 24), and established cell lines (e.g., NK-92) (25, 26). This diverse sourcing supports the development of “off-the-shelf” CAR-NK products, theoretically reducing manufacturing costs and overcoming the limitation of autologous cell unavailability for some cancer patients (27).
CAR-NK also exhibits a more favorable safety profile and robust therapeutic efficacy. Specifically, it carries a lower risk of CRS, immune effector cell-associated neurotoxicity syndrome (ICANS), and GvHD. Notably, CAR-NK can recognize and eliminate tumor cells lacking major histocompatibility complex (MHC) expression (28), a feature that reduces GvHD risk and facilitates allogeneic transplantation. This safety advantage stems from differential cytokine secretion, as CAR-NK primarily secretes interferon (IFN)-γ and granulocyte-macrophage colony-stimulating factor (GM-CSF) (29), whereas CAR-T produces pro-inflammatory cytokines interleukin (IL)-1, IL-6, IL-10, tumor necrosis factor (TNF)-α, monocyte chemoattractant protein-1 (MCP-1) linked to CRS and severe neurotoxicity (30).
Endogenously, NK cells distinguish malignant cells via surface receptors [e.g., activating/inhibitory killer cell immunoglobulin-like receptors (KIRs)] (31), enabling “self-non-self” discrimination and avoiding off-tumor cytotoxicity against normal cells (32). CAR-NK eliminates tumor cells through two key mechanisms, intrinsic cytotoxicity via granzyme and perforin release (33, 34), and antibody-dependent cellular cytotoxicity ADCC (35). The workflow of adoptive CAR-NK therapy for cancer is illustrated in Figure 1.
Schematic diagram illustrating the preparation and clinical application of CAR-NK derived from multiple cellular sources, including peripheral blood mononuclear cells (PBMCs), UCB, iPSCs, and the NK-92 cell line.
Collectively, the accessibility of multiple cellular sources, superior safety profile, and diverse tumor-eliminating mechanisms of CAR-NK overcome the critical barriers of autologous CAR-T and expedite translational progress from preclinical evaluation to clinical application.
Scope and novelty of this review
1.3
This review focuses on CAR-NK, a rapidly evolving field with distinct advantages over CAR-T. Its novel scope encompasses four core aspects.
First, Synthesis of research advances through 2025, including preclinical breakthroughs and ongoing clinical trials for hematological malignancies and solid tumors.
Second, highlights of NK-specific CAR engineering innovations, such as Fc-binding domain modification (CD16 optimization) (36–39), cytokine armoring (membrane-bound IL-15 expression) (40–42), NK-specific co-stimulatory domain incorporation (DNAM1, 2B4) (43–45), and TME-resistant engineering (TGF-β receptor knockout, CXCR1/CXCR4 overexpression) (46–50) to overcome solid tumor barriers.
Third, systematic summary of preclinical and clinical progress in major solid tumors (18, 39, 51–53), e.g., hepatocellular carcinoma, ovarian cancer, gastric cancer, brain tumors, prostate cancer, triple-negative breast cancer, as well as innovative combination therapies, CAR-NK combined with oncolytic viruses (54) or photosensitizing agents, and 3D tumor model validations (17, 55).
Fourth, proposal of actionable solutions to key technical bottlenecks includes autocrine IL-15 engineering NKX101, FT576 and suicide switch design for in vivo persistence, metabolic reprogramming, chemokine receptor modification and dual-target CAR construction for TME suppression, and feeder-cell-free expansion and non-viral transduction CRISPR/Cas9 combined with transposons for industrialization (56, 57).
This review synthesizes cutting-edge research and bridges the translational gap between basic science and clinical practice, serving as a comprehensive reference for researchers and clinicians in the field.
NK cell biology and sources
2
NK cells, a critical component of the innate immune system, play a pivotal role in tumor immunosurveillance and antiviral defense. Their unique biological traits and diverse cellular sources form the foundation for advancing CAR-NK therapy.
Biological characteristics of NK cells
2.1
NK cells are large granular lymphocytes derived from bone marrow hematopoietic stem cells (HSCs). Their maturation follows a sequential differentiation pathway, as HSCs differentiate into NK precursor cells, which further develop into immature NK cells and ultimately into functional mature NK cells (58). This process is regulated by a cytokine cascade including IL-15, IL-7, and stem cell factor (SCF) (19, 58).
In terms of tissue distribution, NK cells primarily reside in PB, accounting for 5%–20% of total lymphocytes (59). They also populate various lymphoid and non-lymphoid tissues (e.g., lung, liver, spleen, bone marrow, lymph nodes) and exhibit tissue-specific functional properties (60, 61). Human NK cells are divided into two major subsets based on CD56 expression levels, namely CD56^bright^ and CD56^dim^. The CD56^bright^ subset, representing an early maturation stage, localizes mainly in secondary lymphoid organs and exerts immunomodulatory effects via secreting cytokines such as IFN-γ, tumor TNF, and GM-CSF (62–64). In contrast, the CD56^dim^ subset predominates in PB and spleen, characterized by potent cytotoxicity against tumor and infected cells. This subset highly expresses CD16 (FcγRIIIA), the receptor mediating ADCC (62–64).
NK cell cytotoxicity is tightly regulated by a balance between activating and inhibitory receptors. Activating receptors (e.g., NKG2D, NKp30, NKp44, NKp46) recognize stress-induced ligands on tumor or infected cells to trigger cytotoxicity. Inhibitory receptors (e.g., KIRs, NKG2A, CD94) bind to major MHC class I molecules on normal cells to prevent off-tumor cytotoxicity (31, 32). This dual-receptor system enables NK cells to distinguish self from non-self without prior antigen sensitization, a key feature differentiating them from adaptive immune cells such as T cells (65, 66).
Sources of NK cells for CAR-NK
2.2
A core advantage of CAR-NK is the availability of multiple cellular sources, which facilitates the development of off-the-shelf therapeutic products.
PB-derived NK cells
2.2.1
PB from healthy donors is a widely used NK cell source. PB-derived NK cells (PB-NK) can be isolated via density gradient centrifugation followed by depletion of T cells, B cells, and monocytes (21, 22). They exhibit robust cytotoxicity and functional stability, making them suitable for autologous or allogeneic transplantation. However, PB-NK cells are limited by donor-dependent expansion variability and the requirement for large blood volumes to achieve clinical-scale production (19, 31).
UCB-derived NK cells
2.2.2
UCB is a rich source of immature NK cells with high proliferative potential and low immunogenicity. UCB-derived NK cells (UCB-NK) can be efficiently expanded ex vivo using cytokine cocktails or feeder cells and display cytotoxicity comparable to PB-NK cells (23). Additionally, UCB is readily accessible from cord blood banks, eliminating the need for donor recruitment and simplifying manufacturing workflows (19, 23). A key limitation is the low NK cell yield per unit volume of UCB, which necessitates efficient expansion strategies to reach therapeutic doses.
iPSC-derived NK cells
2.2.3
iPSCs provide a renewable, standardized source of NK cells. Stepwise cytokine and growth factor induction enables iPSCs to differentiate into NK cells (iPSC-NK), supporting large-scale production of homogeneous cell populations (22, 24, 25). iPSC-NK cells exhibit consistent phenotypic and functional traits across batches, overcoming the donor variability associated with PB-NK and CB-NK cells. Moreover, genetic modifications (e.g., HLA-E knockout, CD47 blockade) can be introduced at the iPSC stage to enhance tumor targeting and reduce immune rejection (22, 37). A critical challenge is the incomplete maturation of iPSC-NK cells, which may lead to impaired in vivo cytotoxicity (19, 67).
NK cell lines
2.2.4
Established NK cell lines (e.g., NK-92, NK-92MI, YT) are extensively used in preclinical and clinical research. NK-92 cells, derived from a non-Hodgkin lymphoma patient, can be indefinitely expanded in vitro and exert potent cytotoxicity against a broad spectrum of tumor cells (26, 27). They are amenable to genetic modification and large-scale production, making them ideal candidates for off-the-shelf therapy. However, NK-92 cells lack MHC class I expression, which may increase immune rejection risk, and clinical application therefore typically requires irradiation to abrogate in vivo proliferation (26, 31).
CAR structure and engineering strategies
3
Basic components and evolution of CARs
3.1
CARs are synthetic fusion proteins typically composed of four core elements, an extracellular domain (ECD), a hinge region, a transmembrane domain (TMD), and an intracellular domain (ICD). As the antigen-binding domain (ABD), the ECD mediates MHC-unrestricted recognition of tumor-specific antigens. Positioned extracellularly, the hinge region acts as a flexible linker between the ECD and TMD, regulating the spatial orientation and overall conformation of the CAR molecule to directly impact antigen-binding efficacy.
The TMD anchors the receptor in the plasma membrane, with sequences often derived from CD3ζ, CD4, CD8, or CD28, components also found in the ICD. Emerging evidence indicates that TMD composition influences CAR expression, molecular stability, immune synapse formation, and even endogenous signal dimerization (67). The ICD is critical for downstream signaling, usually containing both a co-stimulatory domain (from the CD28 family, e.g., CD28, ICOS, or the TNF receptor family, e.g., 4-1BB, OX40, CD27) and a primary signaling domain. These two components synergize to generate robust intracellular activation signals (67).
Research progress in CAR structure design
3.2
Advancements in CAR engineering have spurred the development of multiple generations of CAR-based immunotherapies, with CAR-T serving as a representative model. Structurally, CAR molecules have evolved through at least four generations (Figure 2).
Evolution of CAR generations. First-generation CARs comprise an extracellular antigen-binding domain, a transmembrane domain, and a CD3ζ signaling domain but lack co-stimulatory signals, leading to limited persistence and efficacy. Second-generation CARs integrate a single co-stimulatory domain to provide secondary activation signals, enhancing T cell activation, proliferation, and cytotoxicity. Third-generation CARs incorporate two co-stimulatory domains that act synergistically to further improve T cell persistence and memory formation capacity. Fourth-generation CARs feature a novel “Zip” functional module enabling cytokine secretion or immune cell recruitment, thereby achieving multifunctional expansion and precise therapeutic regulation.
First-generation CARs contain an extracellular antigen-binding domain, a transmembrane domain, and a CD3ζ signaling domain but lack co-stimulatory elements, which renders engineered T cells highly prone to exhaustion. Second-generation CARs integrate a single co-stimulatory domain (e.g., 4-1BB/CD137), enhancing T cell persistence. Notably, most currently approved CAR-T products adopt the second-generation structure (68–70). Third-generation CARs incorporate two co-stimulatory domains (e.g., CD28/4-1BB or CD28/OX40) to prolong cell survival and function, and they are currently under investigation primarily in solid tumor models. Despite these improvements, key challenges remain, including solid tumor targeting, specific antigen recognition, and resistance within the TME. Fourth-generation CARs feature structural innovations such as logic gates, adaptor-dependent circuitry, pharmacological switches, and cytokine secretion modules. Logic gate CARs leverage signal integration from multiple activation or inhibition antigens to improve target recognition and address tumor heterogeneity, for example, dual-target CD19/CD22 CAR-T cells can eliminate cancer cells expressing either antigen (71). Adaptor-dependent CARs rely on supplemental ligands to recognize one or more tumor-associated antigens (TAAs). A bispecific anti-fluorescein isothiocyanate (FITC) adaptor, for instance, bridges CAR-T and tumor cells to induce tumor destruction (72). Additionally, switchable CARs (e.g., those containing the FKBP12 F36V component) allow external regulation of activity via drug administration (73). Cytokine secretion modules are particularly prevalent in CAR-NK, with interleukin-15 (IL-15) integration being a common strategy to enhance NK cell persistence in vivo.
Optimization of CAR structures for CAR-NK
3.3
Current CAR designs for CAR-NK are largely derived from canonical CAR-T constructs, as both share structural and functional similarities in activation domains (e.g., inclusion of CD3ζ and co-stimulatory regions). However, fundamental differences exist between NK cell and T/B cell activation pathways. Emerging research suggests that customizing CARs with NK cell-specific intracellular domains may further enhance anti-tumor activity and overall therapeutic efficacy (43) (Figure 3).
Schematic illustration of CAR-NK receptor architecture, efficacy-enhancing strategies, TME modulation, and induced tumor cell cytotoxicity. (A) CAR-NK receptor architecture: The CAR consists of four modules (1): Targeting Domain (2): Transmembrane Domain (3): Co-Stimulatory Domains (4): Signal Transduction Domain. (B) Enhancement strategies for CAR-NK efficacy. (1): Fc binding proteins + IL15 (2): TGF-β receptor dominant-negative receptor (DNR) (3): CXCR1 receptor. (C) TME modulation by CAR-NK. In the TGF-β-rich immunosuppressive TME, CAR-NK mediates TME remodeling via CXCR1-dependent infiltration and DNR-mediated TGF-β neutralization, while targeting B7H3/CD30 on malignant tumor cells. (D) Cytotoxicity and tumor cell death.
Fc-binding proteins and cytokines
3.3.1
CD16 (FcγRIIIA), a low-affinity IgG receptor expressed on the NK cell surface, plays a pivotal role in mediating ADCC. Incorporating Fc receptor elements into CAR designs has thus become a major research focus. For example, expressing AFM13, a bispecific antibody targeting CD30 and CD16, in UCB-NK cell lines enhances lysis of CD30-positive tumors (36). Dan Kaufman’s group demonstrated that CD16 integration improves the functional performance of iPSC-NK cells (37), while Meng et al. developed a hnCD16-2B4-DAP10-CD3ζ CAR to drive robust ADCC activation (38). Fate Therapeutics has similarly optimized iPSC-NK cell activity using a high-affinity CD16a variant, and Zhang et al. engineered CD33/CD16 CAR-NK with transgenic co-expression of anti-CD16 antibodies to augment cytotoxicity against acute myeloid leukemia (AML) (39).
The in vivo longevity and functional activity of CAR-NK can also be enhanced by engineering specific cytokine expression. Mouse models of MM and AML have shown that transgenic IL-15 expression in CAR constructs not only strengthens anti-tumor effects but also extends NK cell survival (40, 41). Furthermore, metabolic adaptability loss is linked to post-therapy resistance, and combining IL-15 incorporation into the CAR backbone with multiple infusion regimens has been shown to overcome such resistance mechanisms (42).
Co-stimulatory and signal transduction domains
3.3.2
Early engineered CARs were designed to mimic T cell receptor signaling by coupling extracellular binding domains to the TCRζ signaling chain. CAR-NK has largely adopted the first four generations of CAR-T, which typically include canonical CD3ζ, 4-1BB, and CD28 co-stimulatory domains. However, comparative studies of five CAR architectures (CD3ζ, CD28-CD3ζ, DNAM1-CD3ζ, 2B4-CD3ζ, DNAM1-2B4-CD3ζ, with the latter two containing NK cell-specific co-stimulatory signals) revealed that CAR-NK-92 cells engineered with NK cell-specific domains (DNAM1 and 2B4) exhibit significantly higher anti-tumor cytotoxicity than those using T cell-derived co-stimulatory domains (43). Among these designs, dual DNAM1/2B4 CAR-NK-92 cells demonstrated the most potent activity (44). Additionally, CAR designs incorporating the DAP10 co-stimulatory motif have also shown enhanced cytotoxic properties (45).
Tumor infiltration and inhibition of TME
3.3.3
A critical challenge for adoptive cell therapy, especially in solid tumors, is achieving efficient tumor infiltration and counteracting the immunosuppressive TME. Overexpressing chemokine receptors such as CXCR4 can enhance the tumor-homing capacity of CAR-NK (46). In peritoneal ovarian cancer (OC) mouse models, CXCR1 upregulation significantly improved CAR-NK infiltration into tumor tissues without compromising cytotoxicity, ultimately boosting anti-tumor efficacy (47).
The TME plays a central role in regulating tumor growth, metastasis, and therapeutic response. Soluble immunosuppressive factors secreted in the TME can directly or indirectly impair NK cell maturation, proliferation, and effector functions, either by acting on NK cells themselves or by modulating the activity of accessory cells such as antigen-presenting cells (APCs). In patient-derived xenograft (PDX) models of glioblastoma multiforme (GBM), TGF-β blockade prevented NK cell dysfunction induced by glioma stem cells (GSCs), enabling superior tumor control (48). To overcome TME-mediated suppression, Chaudhry et al. engineered NK cells co-expressing a B7H3 CAR and a dominant-negative TGF-β receptor (DNR) in vitro studies showed that these DNR/B7H3-CAR-NK cells effectively targeted refractory central nervous system tumors including GBM (49). Recent work by Ben-Shmuel et al. further identified cancer-associated fibroblasts (CAFs) as key mediators of TME-driven immune evasion, uncovering novel CAF-dependent mechanisms that inhibit NK cell anti-tumor cytotoxicity (50).
Preclinical research
4
CAR-NK in hematological malignancies
4.1
Hematological malignancies encompass aggressive diseases (e.g., leukemias, MM, malignant lymphomas) characterized by high lethality, complex treatment regimens, and poor prognoses. Given their prevalence and adverse outcomes, CAR-NK for hematological cancers has become a focal point of intensive research and innovation (51). This section synthesizes preclinical data to analyze core optimization strategies and technical advances across four key dimensions, such as cell source platforms, target selection and optimization, IL-15 utilization, and therapeutic outcome trends (Table 2).
Cell source platforms
4.1.1
iPSC-NK and UCB-NK cells exhibit high expansion efficiency and stable tumoricidal activity, making them promising candidates for allogeneic “off-the-shelf” therapies. For example, iPSC-NK cells engineered with HLA-E knockout and CD47 blockade show enhanced evasion of host immune rejection while preserving cytotoxicity (74). As a standardized cell line, NK-92 cells enable mass production but traditionally require preclinical irradiation, limiting their utility. A study reduced this requirement via CRISPR/Cas9-mediated CD56 upregulation, expanding their applicability (75). PB-NK cells are easily accessible but display significant interindividual variability in expansion and functional stability. Ex vivo priming with IL-12/IL-18 plus feeder cell co-culture reduces this heterogeneity, improving expansion efficiency by 2.3-fold compared to conventional methods (76).
Target selection and optimization strategies
4.1.2
Validated single-target CAR-NK therapies include CD123 and CD33 for acute myeloid leukemia (AML), BCMA for MM, and CD19 for lymphoma. Notably, CD123-CAR-NK exhibits anti-leukemic efficacy comparable to CAR-T in human hematopoietic stem cell-transplanted AML mouse models, with lower acute toxicity (53). Dual-target strategies represent a critical breakthrough for improving efficacy and reducing recurrence. BCMA/GPRC5D dual-target CAR-NK enhances MM cell killing and reduces tumor escape (42, 77). CD33/CD16 bifunctional CAR-NK achieves an 80% killing rate against AML cells, outperforming single-target regimens (39). Additionally, immune checkpoint knockout (e.g., CRISPR/Cas9-mediated NKG2A deletion) relieves tumor-induced immunosuppression, significantly improving in vitro tumoricidal activity and in vivo anti-tumor function (52, 77).
IL-15 utilization methods
4.1.3
IL-15 is a core cytokine for sustaining CAR-NK persistence and activity, with membrane-bound IL-15 (mbIL-15) outperforming exogenous recombinant IL-15. For example, Nkarta’s NKX101 (NKG2D-CAR-NK) uses mbIL-15 fused to a CD8α hinge domain to enhance surface retention, extending anti-tumor activity to 12 weeks in AML models (56, 78, 79). Co-expression of mbIL-15 and IL-21 in BCMA-CAR-NK promotes memory-like NK cell formation, with 60% of treated mice remaining tumor-free at 6 months (80, 81). IL-15/IL-15Rα sushi domain fusion also reduces systemic cytokine release while maintaining autocrine signaling (82, 83).
Outcome trends
4.1.4
Combined strategies (dual-target design, immune checkpoint knockout, mbIL-15 modification) exhibit synergistic effects. For example, triple-modified (BCMA/GPRC5D + NKG2A knockout + mbIL-15) CAR-NK cells achieve 90% tumor clearance in MM models, compared to 55% with single-modified cells (42, 84). Allogeneic platforms (iPSC-NK, CB-NK, NK-92) are core future directions, as they maintain efficacy while meeting “off-the-shelf” clinical demands. CRISPR-edited iPSC-NK cells (CD38 and CD47 knockout) avoid daratumumab-induced fratricide, enabling combination with standard-of-care drugs (85). Compared to CAR-T, CD19-targeted CAR-NK for B-ALL reduces CRS by 80% (86).
CAR-NK cell therapy in solid tumors
4.2
Solid tumor immunotherapy faces critical challenges, including profound TME immunosuppression and a lack of specific TAAs. Robust preclinical research is positioning CAR-NK as a safer, more efficacious alternative, with mechanistic insights driving innovation. This section analyzes advances across cell source platforms, target selection, TME breakthrough strategies, and IL-15 application, alongside outcome trends and innovative directions (Table 2).
Cell source platforms
4.2.1
NK-92 cells are the most widely used in solid tumor preclinical studies due to their standardization. For example, GPC3-CAR-NK-92 achieves 65% tumor regression in hepatocellular carcinoma (HCC) PDX models (87). MSLN-CAR-NK-92 enhances OC infiltration via matrix metalloproteinase (MMP) secretion (88, 89). iPSC-NK cells engineered with CXCR4 and CD137 co-stimulation show a 3-fold improvement in blood-brain barrier penetration in GBM models (90). PB-NK cells modified with c-MET-CAR and IL-15 exhibit 70% cytotoxicity against c-MET-high NSCLC cells, though interindividual variability persists (91). Placental HSC-derived CYNK-101 (high-affinity CD16) shows 40% longer persistence in gastric cancer models than UCB-NK (92–94).
Target selection and antigen heterogeneity mitigation
4.2.2
Core targets for solid tumors are increasingly well-defined. CD147/GPC3 dual-target CAR-NK reduces antigen escape by 50% in HCC PDX models (95). CLDN6-CAR-NK exhibits 85% cytotoxicity against CLDN6 positive OC cells with no cross-reactivity to normal tissues (96, 97). B7-H3/EGFRvIII dual-target CAR-NK achieves 70% tumor shrinkage in intracranial GBM models (98). CAR-T-validated targets (e.g., NKG2D ligands, FOLR1) are also being translated to CAR-NK, as FOLR1-CAR-NK shows 60% cytotoxicity in OC, comparable to FOLR1-CAR-T but with lower toxicity (99).
IL-15 application and persistence enhancement
4.2.3
Beyond mbIL-15 modification, IL-2Rβγ agonists (e.g., Neo-2/15) combined with CAR-NK represent a novel direction. For example, Neo-2/15-MSLN-CAR-NK exhibits superior proliferation and cytotoxicity in OC cell lines and patient-derived organoids, with reduced immunosuppressive factor secretion and improved survival in animal models (100). ALT-803 (IL-15/IL-15Rα fusion) combined with MSLN-CAR-NK improves proliferation by 3.5-fold in pancreatic cancer models (101). Oncolytic viruses (e.g., OV-IL15C, which expresses an IL-15/IL-15R fusion protein) also enhance CAR-NK infiltration and persistence in brain tumors via TME remodeling and sustained IL-15 delivery (54).
TME breakthrough strategies and outcome trends
4.2.4
Chemokine receptor engineering is a key strategy to overcome TME barriers, and CXCR4-overexpressing EGFRvIII-CAR-NK increases GBM tumor accumulation by 4-fold (102). CCR2b-modified MSLN-CAR-NK improves migration to pancreatic cancer stroma (103). TME modulation also includes adenosine pathway blockade, as CAR-NK secreting a CD73-inhibiting scFv elevate intratumoral ATP levels by 60%, reversing immunosuppression in HCC (104). Combination therapies are expanding rapidly, including anti-VEGF (bevacizumab) combined with CAR-NK in OC, which reduces vascular density and improves tumor penetration (105). CAR-NK paired with PD-L1 inhibitors, low-dose chemotherapy, or oncolytic viruses to enhance tumoricidal effects. Stroma-targeting strategies (e.g., against cancer-associated fibroblasts) to indirectly improve TME conditions. Therapeutic outcomes show CAR-NK is most effective in early-stage solid tumor models, as HER2-CAR-NK eliminates 95% of small-volume gastric cancer xenografts, and combination with low-dose paclitaxel extends efficacy to large tumors (60% shrinkage) (106). However, efficacy in large-volume tumors is limited by poor infiltration, suggesting CAR-NK is optimal for early intervention or combined debulking therapy. Promising advances also include PSMA-CAR-NK plus PD-L1 inhibitors for prostate cancer and CD44v6-CAR-NK for triple-negative breast cancer. Advanced technologies (photosensitizer-loaded NK cell drug delivery, 3D tumor spheroids, multiphoton imaging) provide new tools to overcome TME suppression and enable real-time evaluation of CAR-NK behavior (107, 108).
Comparison of CAR-NK strategies between hematological malignancies and solid tumors
4.3
Preclinical CAR-NK development reveals fundamental strategic differences dictated by the distinct biological contexts of hematological malignancies and solid tumors. These differences stem from their core challenges, namely the efficient eradication of disseminated, accessible tumor cells in hematological malignancies versus overcoming TME physical and immunosuppressive barriers to reach targets in solid tumors.
In hematological malignancies, tumor cells circulate in the blood or reside in the bone marrow, making them directly accessible to intravenously infused CAR-NK. Research thus focuses on maximizing effector cell potency and durability, with key strategies including dual-targeting CARs (e.g., BCMA/GPRC5D in MM) (42, 84) to mitigate antigen escape and immune checkpoint knockout (e.g., NKG2A in AML) to amplify activation. “Cytokine armoring” (e.g., mbIL-15 in NKX101) to ensure long-term persistence (56, 78, 79). The goal is to create a potent, persistent “living drug” for systemic eradication of disseminated disease.
Solid tumors present a “fortress-like” challenge, with malignant cells embedded in a TME that excludes and suppresses immune cells. Research priorities shift to navigation and survival in a hostile niche, with key advances including chemokine receptor engineering (e.g., CXCR4 in GBM) (102) to improve tumor homing. Secretion of neutralizing agents (e.g., CD73 scFv in HCC) (104) to resist immunosuppression. Resistance engineering to soluble inhibitors (e.g., TGF-β in HCC) (49). Bispecific CARs and TME-modifying combinations (e.g., anti-angiogenics, oncolytic viruses) to address antigen heterogeneity and physical barriers.
In summary, hematological malignancy strategies center on enhancing intrinsic CAR-NK capabilities for systemic eradication, while solid tumor strategies focus on engineering CAR-NK to overcome extrinsic TME suppression for localized attack. This distinction in challenges and solutions defines the divergent developmental pathways of CAR-NK across cancer types.
Clinical progress
5
Compelling preclinical outcomes of CAR-NK research have accelerated the translation of this platform into clinical evaluation.
Hematological malignancies
5.1
CAR-NK has shown substantial promise in hematological malignancies, with clinical trials targeting diverse antigens and utilizing multiple NK cell sources (Table 3).
Core target antigens and efficacy breakthroughs
5.1.1
CD19
5.1.1.1
As a well-established target in B-cell malignancies, CD19-directed CAR-NK have demonstrated robust safety and efficacy. A Phase I/II trial of IL-15-secreting CD19-CAR-NK (CAR19-IL-15 NK) in 37 CD19-positive patients achieved a 48.6% objective response rate (ORR) at day 30 without severe toxicities, validating the synergy of CAR targeting and cytokine enhancement (109, 110). This is further supported by 2023 long-term follow-up data (111), which reported a 3-year progression-free survival (PFS) rate of 32.4% in R/R NHL. Globally, ongoing trials include a Chinese Phase II study (NCT04796688) investigating cord blood-derived CD19-CAR-NK, with 2024 interim data (112) showing a 56.7% ORR and median PFS of 14 months. Additionally, 2021 U.S. Phase I data (113) demonstrated that combining CD19-CAR-NK with lenalidomide achieved a 63% ORR in high-risk chronic lymphocytic leukemia (CLL), highlighting the potential of synergistic regimens. Collectively, these studies underscore global efforts to optimize CD19-CAR-NK dosing, patient selection, and combination strategies.
BCMA
5.1.1.2
For R/R MM, BCMA-targeted CAR-NK products have emerged as promising off-the-shelf options. Fate Therapeutics’ iPSC-derived FT576 (BCMA-CAR-NK) exhibited excellent tolerability and efficacy in 2024 Phase I data (114) involving 18 R/R MM patients, achieving a 72.2% ORR, including 38.9% complete response (CR), with no CRS, ICANS, or GvHD. Parallel early-phase trials in China are expanding access to this therapy. For example, a Phase I trial (NCT05008536) of CB-NK-derived BCMA-CAR-NK (115) reported a 58.3% ORR in 12 patients with a median response duration of 11 months. Combination strategies are also under active exploration. A 2022 Phase I study (116) combining BCMA-CAR-NK with the anti-CD38 monoclonal antibody daratumumab achieved an 83% ORR, primarily by enhancing ADCC. These data collectively highlight the potent, well-tolerated profile of BCMA-CAR-NK across platforms and their enhanced efficacy in rational combinations.
CD33
5.1.1.3
Targeting the broadly expressed AML-associated antigen CD33, a 2021 Phase I trial (117) in 10 R/R AML patients confirmed preliminary efficacy and safety, achieving a 40% ORR (including 2 CRs) and addressing an unmet need in this difficult-to-treat population. Ongoing trials are building on this by exploring dual-target strategies to reduce relapse risk. For instance, a Chinese trial (NCT05215015) targeting CD33 and CLL1 reported 2024 interim data (118) showing a 50% ORR in 16 patients and a lower relapse rate (12.5%) compared to historical single-target trials (33%). Combination approaches with standard therapies are also under investigation. A U.S. Phase I trial (NCT04623944) evaluating CD33-CAR-NK plus the hypomethylating agent azacitidine reported 2023 data (119) demonstrating a 57% ORR in elderly AML patients. These developments reflect evolving strategies to enhance the efficacy and durability of CAR-NK for AML.
CD7
5.1.1.4
CD7-CAR-NK trials, focused on T-cell malignancies and AML, have shown promising safety and efficacy in early-phase testing. A Chinese Phase I trial (NCT02742727) using NK-92-derived CD7-CAR-NK cells reported 2022 follow-up data (120) of a 45% ORR in T-cell malignancies, with no fratricide observed due to prior CD7 knockout. This approach leverages NK cells’ inherent resistance to self-targeting to overcome this key challenge. Beyond T-cell malignancies, CD7 is also being explored in AML. A 2024 Phase I study (121) of PB-NK-derived CD7-CAR-NK with IL-15 co-expression reported a 38.9% ORR, suggesting enhanced persistence and expanded therapeutic potential.
NK cell source optimization
5.1.2
iPSC-derived NK cells
5.1.2.1
iPSC-NK cells represent a transformative off-the-shelf platform, enabling large-scale production and broad patient access beyond traditional cell sources. Examples include FT576 (BCMA-targeting) for MM (56, 57, 78) and FT555, which enhances anti-tumor activity via dual GPRC5D/CD38 targeting (often combined with daratumumab). The portfolio expanded further with FT596, a CD19/CD22 dual-targeting candidate whose 2023 Phase I data (122) demonstrated a 66.7% ORR in B-ALL.
The utility of this platform extends beyond oncology. The U.S. trials are investigating iPSC-derived CD19-CAR-NK cells for autoimmune diseases, with 2024 interim data (123) showing reduced pathogenic B cells in SLE patients (NCT06255028). A key innovation improving the durability and repeat dosing potential of these allogeneic products is HLA engineering. For instance, HLA-G expression (124, 125) was shown in a Phase I trial to reduce host immune rejection, enabling successful repeated administrations.
Cord blood and PB-NK cells
5.1.2.2
CB-NK and PB-NK cells remain the most widely used sources in clinical trials, each offering distinct advantages. CB-NK provides an allogeneic, readily available option, while PB-NK enables greater flexibility, such as patient-specific or donor-matched strategies.
Key trials exemplify this utility. CB-NK cells engineered with safety switches and cytokine support have shown improved persistence and controllability. For instance, a trial of CB-NK cells with an inducible caspase-9 (iCasp9) safety switch (NCT03056339) reported 2021 long-term data (126) of a 41.2% 3-year overall survival (OS) in NHL with no severe adverse events. Conversely, PB-NK cells are being actively tested in R/R settings, often via genetic modification. A Phase I study using PB-NK cells modified with an NKG2D-OX40-CD3ζ construct (NCT04623944) showed 2023 results (127) of a 53.8% ORR in R/R AML/MDS. Additionally, donor-matched PB-NK cells have proven valuable post-transplant. A 2024 study (128) demonstrated that their administration following allogeneic hematopoietic stem cell transplantation (allo-HSCT) reduced relapse by 40% compared to HSCT alone.
Safety advantages
5.1.3
A consistent theme across clinical trials is the favorable safety profile of CAR-NK. Major studies have reported no severe CRS, ICANS, or GvHD, making this approach particularly suitable for elderly or frail patients. This safety advantage over CAR-Tis reinforced by a 2025 meta-analysis (129) of 12 Phase I/II trials, which found grade ≥3 CRS in only 0.8% of CAR-NK patients versus 34% in CAR-T trials. Two key engineering strategies further enhance safety. Safety switches, such as iCasp9, were validated in a 2022 Phase I trial (130) that successfully rescued two patients with cytokine release. Concurrently, cytokine modulation, for example mbIL-15, reduces systemic toxicity. Data from 2023 (131) showed that 90% of patients receiving this modification experienced only grade ≤2 adverse events. These features collectively improve controllability and broaden the clinical application potential of CAR-NK.
Solid tumors
5.2
Despite unique challenges, such as TME immunosuppression, antigen heterogeneity, CAR-NK has made meaningful progress in solid tumors, with trials focusing on novel targets and delivery strategies (Table 4).
Target antigens tailored to solid tumor biology
5.2.1
NKG2D-L
5.2.1.1
NKG2D ligands (NKG2D-L) are broadly expressed on various solid tumors, making NKG2D-targeting CAR-NK a promising strategy. Phase I trials across multiple tumor types have demonstrated feasibility. For example, a trial using NKG2D mRNA-engineered NK cells in chemotherapy-refractory metastatic colorectal cancer (CRC) showed successful targeting of metastatic lesions, with a 38.5% ORR and no grade 3/4 CRS (132). Ongoing Chinese trials are exploring this approach for other refractory solid tumors, such as a Phase I study in refractory OC (NCT05213195) reporting a 41.7% disease control rate (DCR) (133). To enhance efficacy, combination strategies with immune checkpoint inhibitors are under investigation. A U.S. Phase I trial combining NKG2D-CAR-NK with the anti-PD-L1 antibody pembrolizumab showed promising interim data (134), achieving a 57% DCR in non-small cell lung cancer (NSCLC).
PD-L1/MUC1
5.2.1.2
Dual-targeting CAR-NK, which combine immune checkpoint modulation and direct tumor antigen targeting, represent a promising strategy to overcome TME immunosuppression. A Phase I trial using PD-1/MUC1-CAR-pNK92 cells in 13 patients with advanced solid tumors (lung, pancreatic, colon, OC) achieved stable disease in 69.2% of patients with no severe CRS or marrow suppression (135). 2024 follow-up data (136) supplemented this profile, reporting a median PFS of 7.2 months with two patients maintaining a partial response (PR) for over 1 year. Additionally, a Phase II trial in pancreatic cancer (NCT04847466) reinforced the mechanism of action, reporting a 53.8% DCR in 2023 (137) and reduced regulatory T cell (Treg) infiltration in tumor biopsies.
ROBO1
5.2.1.3
The translation of ROBO1-targeting strategies for pancreatic ductal adenocarcinoma (PDAC) exemplifies how innovative delivery methods enhance therapeutic efficacy. An initial 2022 case study (138) demonstrated that dual systemic and intratumoral infusion of ROBO1-CAR-NK cells stabilized primary and metastatic tumors for 5 months with only fever as a side effect, directly addressing the poor tumor infiltration limitation of systemic CAR therapy in solid tumors. Building on this proof of concept, a subsequent Phase I trial (NCT03941457) with 15 patients reported 2024 expanded data (139), showing a 46.7% DCR and median PFS of 5.5 months. Notably, the trial highlighted that intratumoral delivery alone achieved a 60% DCR in patients with small-volume tumors (≤5 cm), further supporting the critical role of local administration in overcoming the immunosuppressive PDAC microenvironment.
TROP2/Claudin6
5.2.1.4
Researchers are actively developing CAR-NK targeting emerging tumor-specific antigens (TROP2, Claudin6) for OC, CRC, and pancreatic cancer, aiming to maximize on-target activity while minimizing off-tumor toxicity. Two key ongoing trials exemplify this strategy. A U.S. Phase I trial of CB-NK-derived, IL-15-transduced TROP2-CAR-NK cells for CRC (NCT06358430) reported encouraging 2024 interim data (140), showing a 42.9% ORR with no off-target toxicity. A Chinese Phase I trial investigating PB-NK-derived Claudin6-CAR-NK cells for OC (NCT05410717) demonstrated a 45.5% DCR (including 3 PRs) in 2023 data (141). The advancement of these antigen-specific platforms reflects a focused effort to leverage highly expressed tumor markers for improved safety and efficacy in challenging solid tumors.
Overcoming TME and toxicity challenges
5.2.2
TME modulation
5.2.2.1
TME remodeling strategies are being integrated directly into CAR-NK engineering to enhance activity. A key example is the incorporation of a secreted soluble PD-1 (sPD-1) decoy into the CAR construct. A 2024 Phase I trial for HER2-positive breast cancer (142) showed that sPD-1/HER2-CAR-NK cells achieved a 50% ORR. This efficacy was mediated by dual mechanisms, namely blocking the PD-1/PD-L1 axis to enhance the filtration of both CAR-NK and endogenous CD8^+^ T cells, and this was accomplished with no severe side effects. Another approach directly targets suppressive TME signals. Specifically, knocking out the TGF-β receptor in MSLN-CAR-NK (143) effectively reversed TME-induced NK cell exhaustion. This translated to a clinically significant improvement in NSCLC, with the DCR rising from 40% to 65%.
Safety and tolerability
5.2.2.2
Consistent with hematological trials, CAR-NK for solid tumors maintains a favorable safety profile with minimal severe toxicity. For instance, a clinical study combining Hsp70-activated NK cells with standard chemoradiotherapy for stage III NSCLC (144) achieved a promising 67% 1-year OS rate (doubling the historical 33% benchmark) with no grade ≥3 adverse events, highlighting the potential of integrating cellular therapy with conventional treatment. This low-toxicity trend is further corroborated by a 2024 meta-analysis (145) of solid tumor CAR-NK trials, which reported grade ≥3 toxicity in only 11.3% of patients. Most adverse events were manageable, primarily fever and fatigue.
Global trial trends
5.2.3
China and the U.S. lead the clinical development of CAR-NK for solid tumors with distinct yet complementary focuses. China is conducting Phase I/II trials targeting antigens highly expressed in specific malignancies (ROBO1, Claudin6, DLL3). For example, a DLL3-targeting trial in small cell lung cancer (SCLC, NCT05507593) reported 2024 interim data (146) with a 40% DCR. Meanwhile, the U.S. advances the off-the-shelf approach, pioneering iPSC-derived CAR-NK products for broader application. A Phase I trial in renal cell carcinoma (NCT05703854) showed a 38.5% ORR in 2023 data (147). A prominent cross-regional trend is the shift toward combination therapies to enhance efficacy, exemplified by a Phase II trial combining PSMA-CAR-NK with enzalutamide in prostate cancer, which reported a 57% ORR in 2024 (148). This collaborative, combinatorial landscape underscores global efforts to overcome the unique challenges of solid tumor treatment.
Autoimmune diseases
5.3
Building on oncology success, CAR-NK is expanding into autoimmune disease treatment, leveraging its ability to selectively eliminate pathogenic immune cells while sparing normal tissues. Early-phase trials target B-cell-associated antigens (CD19, BCMA) in conditions like SLE. A Chinese Phase I trial of CD19-CAR-NK for SLE (NCT06010472) reported encouraging 2024 interim data (149), with 66.7% of patients achieving a SLE disease activity index 2000 response (SLEDAI-2K response) and reduced anti-dsDNA antibodies. Similarly, a U.S. trial of iPSC-derived CD19-CAR-NK for refractory autoimmune diseases (NCT06255028) demonstrated a 58.3% DCR in Sjogren’s syndrome in 2023 Phase I results (150). To enhance specificity and efficacy, next-generation constructs are under exploration. An ongoing trial of CD19/BCMA dual-target CAR-NK for SLE (NCT06792799) is supported by 2024 preclinical data (151) showing selective elimination of autoreactive B cells without affecting normal counterparts. This targeted approach offers a potential advantage over traditional broad immunosuppression by directly addressing disease pathology while minimizing systemic toxicity.
Key trends and future directions
5.4
From single to multi-targeting
5.4.1
To overcome tumor antigen heterogeneity and reduce relapse risk, CAR-NK trials increasingly adopt multi-target strategies, combining well-established antigens (CD19/CD22, CD33/CLL1, CD19/BCMA) to broaden anti-tumor responses. This evolution is advancing toward triple-target approaches. For instance, a construct targeting CD19, CD22, and CD79b (NCT06727383) showed remarkable 2024 preclinical efficacy (152), achieving 95% tumor clearance in B-cell malignancy models. Beyond hematological cancers, cross-tumor targeting (e.g., NKG2D-Ligand + MSLN) is being explored to develop platforms effective against a wider spectrum of metastatic solid tumors.
Off-the-shelf scalability
5.4.2
iPSC-derived NK cells are becoming a cornerstone of global CAR-NK trials, providing a standardized, scalable off-the-shelf platform that overcomes logistical and manufacturing limitations of autologous cell therapies. This paradigm shift is exemplified by industrial-scale platforms like Fate Therapeutics’ FT500, which 2023 data (153) showed can reliably produce over 1×10^12^ cells per batch with consistent potency across donors, enabling rapid patient access. Parallel development of robust cryopreservation protocols for other allogeneic sources (e.g., CB-NK) facilitates global distribution. A 2024 study (154) demonstrated that cryopreserved CB-NK cells maintain ~80% viability post-thaw, ensuring functionality upon delivery. These bioprocessing and logistics advances are critical for translating CAR-NK therapeutic potential into widespread clinical access.
TME and cytokine optimization
5.4.3
Beyond foundational IL-15, sPD-1 decoy, or NKG2D enhancements, the next frontier involves engineering CAR-NK as dynamic “TME modifiers” that actively reprogram the tumor ecosystem. For example, CAR-NK engineered to secrete IL-12 (155) in GBM models successfully repolarized tumor-associated macrophages toward an anti-tumor M1 phenotype, enhancing CAR-NK infiltration. To manage risks of potent modifications, sophisticated cytokine control mechanisms are under development. A 2024 study (156) showed that miR-155 knockout provides fine-tuned cytokine release regulation, reducing systemic toxicity while preserving therapeutic function. These strategies represent a shift from arming NK cells to transforming them into intelligent, self-regulating agents capable of reshaping hostile tumor environments.
Expanded disease scope
5.4.4
Beyond oncology, CAR-NK is expanding into new therapeutic frontiers. It offers a targeted alternative to conventional broad-spectrum immunosuppressants in autoimmune diseases, enabling selective elimination of pathogenic cells. Its reach extends further. 2023 preclinical data (157) demonstrated CD20-CAR-NK efficacy against Epstein-Barr virus (EBV)-associated diseases, and future clinical trials plan to target viral hepatitis (HBV) by eliminating HBsAg-expressing hepatocytes, showcasing its potential as a versatile platform for diverse pathological conditions.
CAR-NK has evolved from early-phase safety testing to targeted, multi-modal approaches across diverse diseases. Its favorable safety profile, flexible cell sources, and off-the-shelf potential position it as a transformative therapy, with ongoing trials focusing on optimizing targets, combinations, and delivery to maximize clinical impact.
Comparison of clinical outcomes
5.5
CAR-NK has demonstrated notable clinical potential in both hematological malignancies and solid tumors, yet distinct patterns of efficacy, safety, and therapeutic challenges characterize its application in these two disease categories.
Efficacy and response patterns
5.5.1
CAR-NK exhibits a clear efficacy gradient between hematological malignancies and solid tumors, largely driven by target accessibility and TME characteristics. In hematological malignancies, more established and pronounced efficacy has been achieved, particularly against well-defined lineage antigens (CD19 in B-cell malignancies, BCMA in MM). A Phase I/II CD19-CAR-NK trial reported a 48.6% ORR at day 30, and BCMA-targeting products like FT576 show high tolerability and promising disease control. A 2025 meta-analysis of 28 trials (158) quantifies this advantage, with a median ORR of 52.3% (range: 38.9–72.2%) and median PFS of 11.2 months in hematological cancers, where responses are typically deeper and more durable.
Solid tumors present significant challenges, such as antigen heterogeneity, physical barriers, profound TME immunosuppression, leading to more modest clinical responses often characterized by disease stabilization rather than complete remission. The same 2025 meta-analysis reported a significantly lower median ORR (31.7%, range: 28.5–57.1%) and shorter median PFS (6.8 months) for solid tumors. This profile is exemplified by dual-targeting PD-1/MUC1-CAR-NK trials (69.2% stable disease in advanced solid tumors) and ROBO1-CAR-NK therapy (several months of tumor stabilization in PDAC). This efficacy gap underscores the critical need for innovative strategies to overcome the unique suppressive landscape of solid tumors.
Safety and tolerability
5.5.2
A consistent, defining advantage of CAR-NK across both disease categories is its favorable safety profile. Unlike CAR-T, key studies consistently report no severe CRS, ICANS, or GvHD, making CAR-NK particularly suitable for elderly or frail patients. A 2024 pooled analysis (159) quantifies this, reporting that grade ≥3 CRS occurred in only 0.9% of hematological malignancy patients and 1.2% of solid tumor patients. No GvHD was reported in either group, though solid tumor trials showed a slightly higher rate of manageable injection-site reactions (8% vs. 3% in hematological trials). This consistent safety record underscores a critical therapeutic advantage of the CAR-NK platform.
Key therapeutic challenges
5.5.3
CAR-NK clinical translation faces distinct yet formidable challenges in hematological and solid tumor contexts, requiring tailored solutions.
In hematological malignancies, primary challenges are antigen escape and disease relapse. For instance, 2023 data (160) indicated 25% of B-ALL relapses are due to CD19 loss, spurring dual-target approaches (e.g., CD19/CD22, CD33/CLL1) to broaden antigen coverage. Concurrent efforts focus on optimizing in vivo NK cell persistence and expansion, frequently via cytokine engineering (e.g., constitutive IL-15 secretion).
For solid tumors, barriers are structural and biological, rooted in the hostile TME. A 2024 study (161) quantified this challenge, reporting that TME factors such as TGF-β and adenosine reduce CAR-NK cytotoxicity by 40–60%. Additional hurdles include poor tumor infiltration and antigen heterogeneity. Multi-pronged strategies are being pursued to overcome these challenges. For example, innovative delivery approaches, such as the local or intratumoral administration of ROBO1-CAR-NK, are under investigation. Additionally, CAR-NK is being engineered to actively modify the TME, including through the secretion of sPD-1 decoy. Another key strategy involves targeting broadly expressed antigens, like NKG2D-Ligand, to circumvent tumor antigen heterogeneity.
Divergence in clinical development maturity: hematological vs. solid tumors
5.5.4
CAR-NK clinical development exhibits a clear maturity dichotomy between hematological malignancies and solid tumors, reflecting distinct biological challenges.
In hematological malignancies, translation is more advanced, as evidenced by numerous Phase I/II trials are either completed or recruiting. This progress is paving the way toward late-phase evaluation and commercialization. Three ongoing Phase III trials (targeting CD19, BCMA, CD33) underscore this momentum, and the iPSC-derived FT576 (BCMA) is expected to file for FDA approval in 2025. Current efforts focus on optimizing combination regimens and scaling off-the-shelf platforms for broader access.
In contrast, CAR-NK for solid tumors remains predominantly in early-phase exploratory stages (≈80% Phase I/II). Research focuses on overcoming TME barriers via novel target discovery (Claudin6, DLL3, TROP2), delivery optimization (local/intratumoral administration), and rational combinations with immunotherapy or chemotherapy. Promising targets (TROP2, ROBO1) are advancing into Phase II studies, signaling a maturing pipeline.
In summary, while CAR-NK demonstrates stronger, more immediate efficacy in hematological cancers, successful solid tumor application requires continued innovation in targeting, delivery, and TME modulation. Both arenas benefit greatly from the platform’s consistent safety profile, which supports broader clinical development and provides a foundation for testing increasingly sophisticated therapeutic strategies.
Analysis of current limitations and future prospects of CAR-NK
6
While CAR-NK has emerged as a promising off-the-shelf immunotherapeutic strategy for hematological and solid malignancies, recent clinical and preclinical advances have highlighted persistent challenges hindering its full translational potential. Building on foundational insights from prior work and refined by contemporary studies (17), this discussion focuses on core challenges and integrates the latest evidence and mechanistic insights.
Persistence and in vivo expansion
6.1
The clinical utility of CAR-NK is tightly linked to its ability to achieve sufficient cell quantities for therapeutic efficacy and maintain functional activity in vivo, two interconnected challenges that span ex vivo preparation and in vivo survival.
Ex vivo expansion: balancing quantity, quality, and safety
6.1.1
A primary bottleneck for clinical application is the limited abundance of NK cells (5–15% of peripheral blood lymphocytes) and their inherently low proliferative capacity, requiring robust ex vivo expansion to achieve therapeutic doses (1, 38). While feeder cell-based and cytokine-cocktail approaches have advanced, 2020–2025 studies reveal unresolved trade-offs between scalability, safety, and functional integrity.
Genetically engineered K562 cells (expressing mbIL-15/4-1BBL or mbIL-21) remain the gold standard for expansion, but their tumorigenic origin raises contamination risks in clinical products (109, 162). Recent work by Liu et al. (2020) demonstrated that 721.221-mIL-21 feeder cells enhance CAR-NK expansion by 30-fold while preserving telomere length, though non-specific T cell co-proliferation remains a concern (109). Novel feeder-free systems using artificial antigen-presenting cells (aAPCs) with biodegradable scaffolds show promise, but their clinical compatibility is still under evaluation (42).
Combinations of IL-2, IL-15, and IL-21 have been widely used for expansion, but a 2022 study by Wagner et al. identified that a two-phase protocol (IL-15 priming followed by IL-21 stimulation) improves NK cell cytotoxicity against solid tumors by upregulating NKG2D and CD16 (163). However, cytokine-driven expansion often induces functional exhaustion, as shown by increased TIM-3 expression in expanded NK cells (41, 164).
GMP-compliant expansion of CAR-NK for large-scale clinical use remains challenging. Automated bioreactor systems have improved yield uniformity, but PB-NK cells still exhibit donor-dependent expansion variability (67, 165). iPSC-NK cells offer a homogeneous alternative, yet their immature phenotype and high inhibitory receptor expression limit immediate clinical utility (41, 67).
In vivo persistence: extending survival and function
6.1.2
CAR-NK exhibits a shorter in vivo lifespan (days to weeks) than CAR-T, limiting long-term anti-tumor efficacy (1, 29). Previous studies have focused on cytokine engineering and genetic modification to address this limitation.
Systemic IL-2 or IL-15 administration enhances persistence but carries the risk of cytokine release syndrome (CRS) (175, 176). Membrane-bound IL-15 (mbIL-15) integrated into CAR constructs enables autocrine signaling, extending in vivo survival to >12 weeks in murine models (177). The IL-15 superagonist ALT-803 improves persistence without inducing CRS, though clinical data remain limited (178, 179). TriKE (CD33-bi-specific + IL-15) promotes NK cell expansion in AML patients, with ongoing Phase I trials (NCT03214666) showing promising persistence (180, 181).
Genetic modifications also play a role in enhancing persistence. CRISPR/Cas9-mediated knockout of CISH (a negative regulator of IL-15 signaling) improves CAR-NK persistence by 3-fold in vivo (41, 182). Overexpression of anti-apoptotic genes (BCL-2, MCL-1) reduces NK cell exhaustion, but tumorigenicity concerns restrict clinical application (179). Suicide genes (e.g., iC9) provide safety control but do not directly address persistence (80).
Source-dependent variability further impacts in vivo longevity: UCB-derived CAR-NK exhibits longer persistence than PB-derived cells due to higher proliferation potential, but their immature phenotype impairs immediate cytotoxicity (25, 46). iPSC-derived CAR-NK offers unlimited expansion but require maturation steps to enhance survival in the TME (41, 67).
CAR transduction: enhancing efficiency and stability
6.2
NK cells are inherently resistant to genetic modification, with historically lower transduction efficiencies than T cells. Recent studies have focused on optimizing viral and non-viral methods to improve CAR delivery while minimizing cytotoxicity, as efficient transduction is prerequisite for consistent in vivo function.
Lentiviral vectors remain preferred for stable integration, but their efficiency in primary NK cells typically ranges from 29–50% (54, 166). The addition of phytohaemagglutinin (PHA) or cytokine pre-stimulation (IL-2 and IL-12) increases lentiviral transduction to 80% (54, 167). Adeno-associated virus (AAV) serotype 6 has emerged as a safer alternative, achieving 68% transduction in primary NK cells without insertional mutagenesis (168, 169). Retroviral vectors, while efficient (43–93%), are limited to dividing cells and carry higher mutagenic risk (170, 171).
Non-viral approaches offer alternative pathways: mRNA electroporation provides transient but high-efficiency CAR expression (80–90%), yet its short half-life (3–5 days) and poor performance in UCB-NK cells restrict utility (54, 172). 3D nanochannel-electroporation (NEP) addresses cell membrane damage, achieving 70% transduction with improved viability (172, 173). CRISPR/Cas9-mediated site-specific integration (e.g., into the TRAC locus) enables stable CAR expression, but high costs and technical complexity hinder widespread adoption (168).
Conventional CAR constructs (optimized for T cells) fail to fully activate NK cells. Recent studies highlight the importance of NK-specific co-stimulatory domains (2B4, DAP10, DAP12) and transmembrane domains (NKG2D, NKp46) for enhancing transduction efficiency and function (62, 70). For example, 2B4-containing CARs improve NK cell proliferation and cytokine production by 2.5-fold compared to CD28-containing constructs (174).
Barriers in solid tumors
6.3
The immunosuppressive TME remains the greatest challenge for CAR-NK in solid tumors, inducing NK cell dysfunction, metabolic exhaustion, and impaired trafficking (1, 183). Recent studies (2020–2025) have uncovered novel mechanisms of suppression and potential solutions.
Metabolic dysfunction
6.3.1
Hypoxia and nutrient depletion in the TME downregulate NK cell glycolysis and oxidative phosphorylation, reducing IFN-γ production and cytotoxicity (184, 185). Targeting metabolic checkpoints (e.g., GLUT1 overexpression) improves CAR-NK glucose uptake and function in hypoxic tumors (186). Lactate dehydrogenase (LDH) inhibition reverses TME acidosis, restoring NK cell perforin/granzyme expression (187, 188).
Immunosuppressive signaling
6.3.2
Checkpoint molecules (PD-1, TIGIT, NKG2A) and inhibitory cytokines (TGF-β, IL-10) suppress NK cell activation. PD-1 knockout CAR-NK exhibits 2-fold higher cytotoxicity against PD-L1 positive lung cancer cells (189, 190). Anti-NKG2A antibodies (e.g., monalizumab) block HLA-E binding, enhancing CAR-NK function in glioblastoma models (78, 191). TGF-βRII knockout using CRISPR/Cas9 improves NK cell resistance to TME suppression in AML (192).
Trafficking and infiltration
6.3.3
CAR-NK struggles to penetrate solid tumor masses due to extracellular matrix (ECM) barriers and deficient chemokine signaling. Engineering CAR-NK to express CXCR4 or CXCR1 enhances homing to CXCL12+ or CXCL8+ tumors, increasing intratumoral infiltration by 30% (193). Combination with ECM-degrading enzymes (e.g., hyaluronidase) further improves trafficking (194, 195).
Antigen heterogeneity and loss
6.3.4
Solid tumors often exhibit heterogeneous antigen expression, leading to immune escape. Dual-target CARs (e.g., EGFR/EGFRvIII, GD2/NKG2DL) overcome this by recognizing multiple antigens, reducing relapse rates in glioblastoma and neuroblastoma models (96, 196). Trogocytosis (antigen transfer from tumor to NK cells) also contributes to escape, but blocking CD9 or KIR signaling mitigates this effect (197, 198).
Safety and toxicity profile
6.4
Despite CAR-NK’s favorable overall safety compared to CAR-T, emerging clinical and preclinical data highlight context-dependent toxicities and unresolved safety gaps that require targeted optimization.
Low-grade but prevalent toxicities
6.4.1
Mild to moderate adverse events (AEs) are common, even in the absence of severe CRS or GvHD. Transient fever (40–60% of patients), fatigue (30–45%), and myelosuppression (25–35%) are the most frequently reported AEs across trials (129, 145). These are often linked to cytokine release (e.g., IFN-γ, GM-CSF) or off-tumor targeting of low-expression antigens on normal tissues. For example, CD33-CAR-NK has been associated with transient neutropenia due to CD33 expression on myeloid precursors (117), while MSLN-CAR-NK may cause mild epithelial toxicity in lung or ovarian tissue (88).
Rare but severe safety risks
6.4.2
Severe toxicities, though rare, present critical concerns. Cases of delayed-onset cytokine release syndrome (grade ≥3, <1% of patients) have been reported in high-dose iPSC-NK trials, linked to uncontrolled proliferation of engineered cells (129). Immune-related adverse events (irAEs), such as rash or colitis, are infrequent but have been observed with dual-target CARs (e.g., CD19/CD22) due to cross-reactivity with normal B cells (122). Additionally, NK cell line-derived products (e.g., NK-92) carry a small risk of insertional mutagenesis if not fully irradiated, though clinical data to date show no tumorigenicity (26).
Safety optimization challenges
6.4.3
Key barriers to improving safety include balancing efficacy and toxicity in cytokine engineering. While mbIL-15 enhances persistence, overexpression can lead to excessive NK cell activation and tissue infiltration (131). Similarly, dual-target or multi-engineering strategies (e.g., checkpoint knockout + cytokine armoring) may increase off-tumor activity by amplifying NK cell cytotoxicity beyond tumor-specific limits (84). Standardized toxicity grading for CAR-NK is also lacking, with variability in how AEs (e.g., cytokine release vs. irAEs) are classified across trials, hindering cross-study comparisons.
Manufacturing and regulatory considerations
6.5
The translation of CAR-NK from preclinical development to routine clinical use is heavily constrained by manufacturing complexity and evolving regulatory frameworks. These challenges are distinct from CAR-T due to CAR-NK’s diverse cell sources, off-the-shelf potential, and unique biological properties.
Manufacturing bottlenecks
6.5.1
Standardization across cell sources
6.5.1.1
Heterogeneity in starting materials complicates consistent production. PB-NK and UCB-NK cells exhibit donor-dependent variability in expansion potential and functional activity, requiring extensive donor screening and batch testing (165). While iPSC-NK cells offer standardized, renewable sources, their differentiation into fully mature, functional NK cells remains inefficient—current protocols yield only 30–50% mature NK cells, with the remainder consisting of immature progenitors (24, 67). NK cell lines (e.g., NK-92) provide consistency but require irradiation to prevent in vivo proliferation, which impairs long-term persistence (26).
Scale-up and cost challenges
6.5.1.2
GMP-compliant scale-up of CAR-NK remains costly and logistically demanding. Feeder cell-based expansion systems, while effective, require additional manufacturing steps to ensure feeder cell depletion, increasing production time and risk of contamination (109, 162). Feeder-free systems (e.g., aAPCs, cytokine cocktails) reduce contamination risk but often result in lower expansion yields (42). For off-the-shelf products, cryopreservation is critical, but current protocols can reduce CAR-NK viability by 20–30% post-thaw, particularly for primary NK cells (154). The high cost of viral vectors (lentivirus, AAV) and genetic engineering tools (CRISPR/Cas9) further limits accessibility, with current manufacturing costs per patient exceeding 50,000–$80,000 for allogeneic products.
Quality control complexity
6.5.1.3
QC for CAR-NK requires multifaceted assessment of identity, purity, potency, and safety. Identity testing must confirm CAR expression (typically via flow cytometry) and NK cell phenotype (e.g., CD56+, CD3-). Purity standards demand <5% contaminating T cells to mitigate GvHD risk, though achieving this in PB-NK or UCB-NK preparations remains challenging (21, 22). Potency assays, which measure cytotoxicity against target tumor cells or cytokine secretion (IFN-γ, GM-CSF), lack standardization across laboratories, leading to variable results. Safety QC includes testing for adventitious agents (viruses, bacteria), insertional mutagenesis (for viral transduction), and off-tumor toxicity (e.g., against normal tissues expressing low levels of target antigens).
Regulatory hurdles
6.5.2
Classification and approval pathways
6.5.2.1
Regulatory agencies (FDA, EMA, NMPA) classify CAR-NK as advanced therapy medicinal products (ATMPs), but specific guidelines for NK cell-based therapies remain evolving. Unlike CAR-T, which has well-established approval pathways for hematological malignancies, CAR-NK faces uncertainty due to its off-the-shelf nature and diverse cell sources. For example, iPSC-derived CAR-NK requires additional data on long-term safety (e.g., tumorigenicity, immune rejection) and consistency across large-scale batches (23, 125). Regulatory bodies also require comprehensive preclinical data on in vivo persistence, toxicity, and efficacy in relevant animal models, which can be challenging for solid tumors due to the lack of standardized PDX models (158).
Labeling and risk management
6.5.2.2
Regulatory requirements for labeling and risk management plans (RMPs) are stringent for CAR-NK. Labels must include detailed information on cell source, manufacturing process, storage conditions, and administration guidelines. RMPs must address potential risks such as CRS, ICANS, GvHD (though rare), and delayed adverse events (e.g., immune suppression, secondary malignancies) (129, 145). For allogeneic products, labeling must include donor screening information (e.g., HLA typing, infectious disease testing) to inform clinicians of potential immune compatibility issues (22, 27).
Global regulatory alignment
6.5.2.3
Disparities in regulatory requirements across regions hinder global development. For example, the FDA requires more extensive data on iPSC differentiation and genetic modification than the NMPA, while the EMA emphasizes long-term follow-up of patients receiving allogeneic products. This lack of alignment increases development costs and delays market access. Additionally, emerging technologies such as CRISPR/Cas9-mediated gene editing in CAR-NK require regulatory clarification on safety and efficacy thresholds, as off-target effects remain a concern (168, 192).
Future directions for manufacturing and regulatory advancement
6.5.3
Manufacturing innovations
6.5.3.1
To address standardization and scale-up challenges, researchers are developing automated, closed-system bioreactors that reduce human error and contamination risk (165). Feeder-free expansion systems using synthetic cytokines (e.g., IL-15 superagonists) and aAPCs with defined components are being optimized to improve yield and consistency (42, 178). Non-viral transduction methods (e.g., 3D nanochannel-electroporation, lipid nanoparticles) are being explored to reduce costs and insertional mutagenesis risk (172, 173). Additionally, cryopreservation technologies (e.g., vitrification) are being refined to improve post-thaw viability and function (154).
Regulatory collaboration and guideline development
6.5.3.2
Increased collaboration between regulatory agencies, industry, and academia is critical to developing harmonized guidelines for CAR-NK. Initiatives such as the FDA’s Cellular, Tissue, and Gene Therapies Advisory Committee (CTGTAC) and the EMA’s Committee for Advanced Therapies (CAT) are working to update ATMP guidelines to include specific considerations for NK cell-based therapies. The development of standardized potency assays and preclinical models (e.g., 3D tumor spheroids, humanized mice) will facilitate regulatory review and reduce variability in clinical data (107, 108). Additionally, post-marketing surveillance programs for approved CAR-NK products will provide long-term safety and efficacy data, informing future regulatory decisions (129, 159).
Discussion
7
Compared with CAR-T, CAR-NK exhibits comparable efficacy against hematological malignancies and holds additional promise for solid tumor applications. Preclinical evidence suggests that using NK cells (rather than T cells) for CAR expression may enhance therapeutic efficacy in solid tumors. Abundant cell sources, absence of GvHD risk, and a favorable safety profile render CAR-NK a highly accessible therapeutic platform.
This review comprehensively synthesizes advances in basic research, preclinical validation, and early clinical outcomes, laying a foundation for further translational development. The continued refinement of CAR constructs, combined with targeted improvements in NK cell persistence and cytotoxicity (e.g., optimizing CD16 affinity and immune checkpoint modulation), will be critical to expanding the clinical potential of CAR-NK.
While definitive results from pivotal solid tumor clinical trials are pending, accumulating evidence from preclinical and preliminary clinical studies underscores a robust and promising future for CAR-NK immunotherapy.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Bock TJ Colonne CK Fiorenza S Turtle CJ . Outcome correlates of approved CD 19-targeted CAR T cells for large B cell lymphoma. Nat Rev Clin Oncol. (2025) 22:241–61. doi: 10.1038/s 41571-025-00992-5, PMID: 39966627 · doi ↗ · pubmed ↗
- 2Bouchkouj N Lin X Wang X Przepiorka D Xu Z Purohit-Sheth T . FDA approval summary: brexucabtagene autoleucel for treatment of adults with relapsed or refractory B-cell precursor acute lymphoblastic leukemia. Oncologist. (2022) 27:892–9. doi: 10.1093/oncolo/oyac 163, PMID: 35983953 PMC 9526496 · doi ↗ · pubmed ↗
- 3Hansen DK Sidana S Peres LC Colin Leitzinger C Shune L Shrewsbury A . Idecabtagene vicleucel for relapsed/refractory multiple myeloma: real-world experience from the myeloma CAR T consortium. J Clin Oncol. (2023) 41:2087–97. doi: 10.1200/JCO.22.01365, PMID: 36623248 PMC 10082273 · doi ↗ · pubmed ↗
- 4Ying Z Yang H Guo Y Li W Zou D Zhou D . Relmacabtagene autoleucel (relma-cel) CD 19 CAR-T therapy for adults with heavily pretreated relapsed/refractory large B-cell lymphoma in China. Cancer Med. (2021) 10:999–1011. doi: 10.1002/cam 4.3686, PMID: 33382529 PMC 7897944 · doi ↗ · pubmed ↗
- 5The Medical Letter . Ciltacabtagene autoleucel (Carvykti) for multiple myeloma (2025). Available online at: https://secure.medicalletter.org/TML-article-1663 d (Accessed September 02, 2023).
- 6Keam SJ . Equecabtagene autoleucel: first approval. Mol Diagnosis Ther. (2023) 27:781–7. doi: 10.1007/s 40291-023-00673-y, PMID: 37658205 · doi ↗ · pubmed ↗
- 7Nie T . Talicabtagene autoleucel: first approval. Mol Diagnosis Ther. (2024) 28:495–9. doi: 10.1007/s 40291-024-00719-9, PMID: 38780683 · doi ↗ · pubmed ↗
- 8Dhillon S . Zevorcabtagene autoleucel: first approval. Mol Diagnosis Ther. (2024) 28:501–6. doi: 10.1007/s 40291-024-00723-z, PMID: 38888762 · doi ↗ · pubmed ↗