RNA-sequencing based gene variants observed in patients with hyperlipidemia and premature coronary heart disease: A preliminary study
Wilanee Dechkhajorn, Kriengchai Prasongsukarn, Surachet Benjathummarak, Supachai Topanurak, Yaowapa Maneerat

TL;DR
This study identifies gene variants linked to premature heart disease in patients with high cholesterol and genetic lipid disorders.
Contribution
The study reports novel gene variants (MAFG, AKAP1, TLR5, CHUK, EMC10, PLRG1) potentially associated with FH-CHD development.
Findings
RNA-seq identified 15 shared gene variants between hyperlipidemia and FH-CHD groups.
Six genes showed high-impact variants predicted to affect gene function and contribute to heart disease.
Variants in MAFG, AKAP1, TLR5, CHUK, EMC10, and PLRG1 may be linked to atherogenesis and CHD.
Abstract
Familial hypercholesterolemia (FH) is a genetic disorder characterized by markedly elevated low-density lipoprotein (LDL) cholesterol levels, which primarily progresses to premature or familial coronary heart disease (FH-CHD). This cross-sectional study included healthy controls (N) and patients with hyperlipidemia (H), FH, CHD, and FH-CHD. We attempted to explore gene variants shared in H, FH and FH-CHD using next-generation sequencing tool. The RNA-seq transcriptome profiling from the whole peripheral blood (n = 3/group) were analyzed. The results revealed 15 intersected gene variants between the H/FH and FH-CHD groups. Aligning and mapping on the coding regions showed significant high-impact variants in 6 of the 15 genes including MAFG, AKAP1, TLR5, CHUK, EMC10, and PLRG1. The significant high-impact variations included frameshift variants in CHUK and PLRG, stop-gain variation in…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
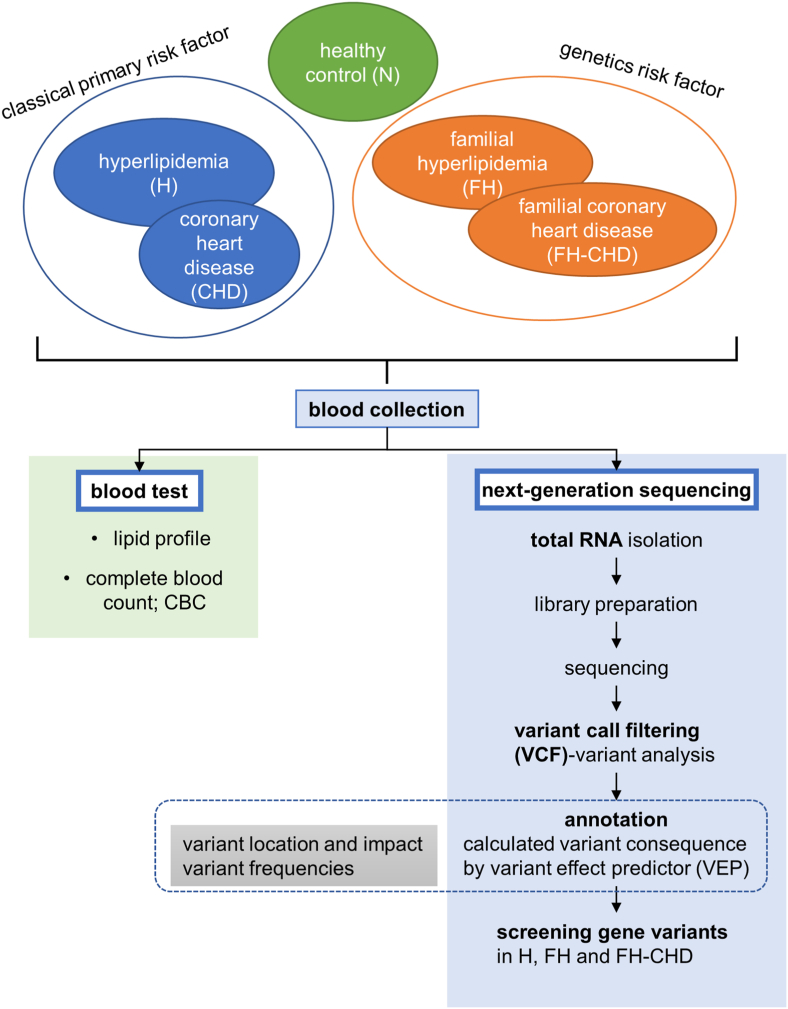 Figure 1
Figure 1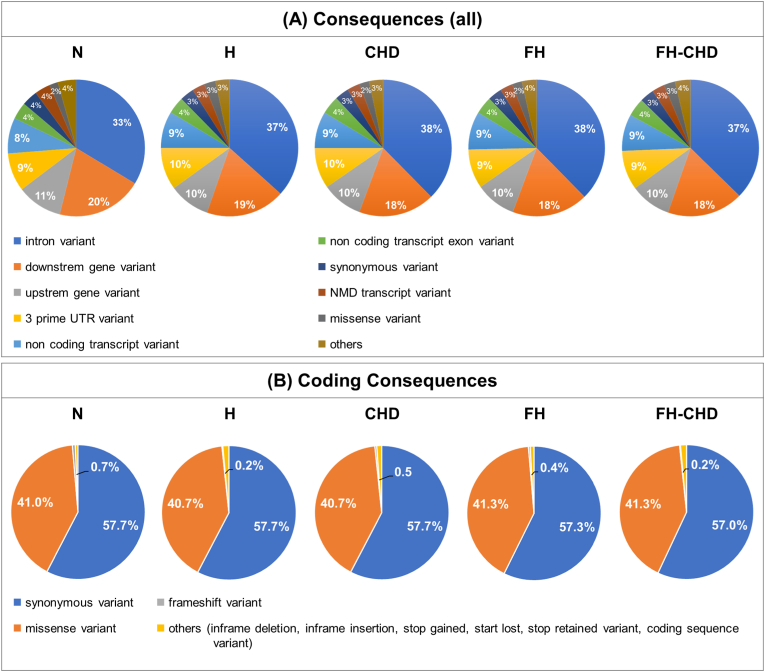 Figure 2
Figure 2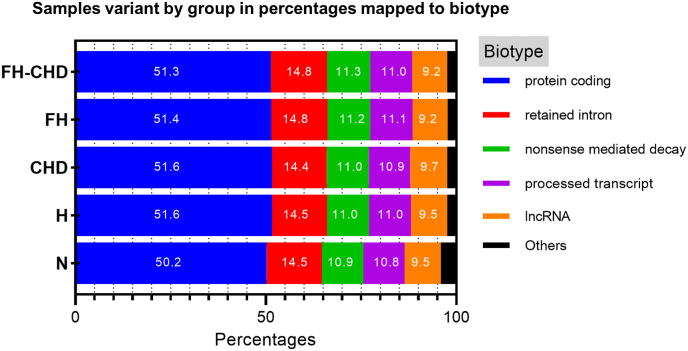 Figure 3
Figure 3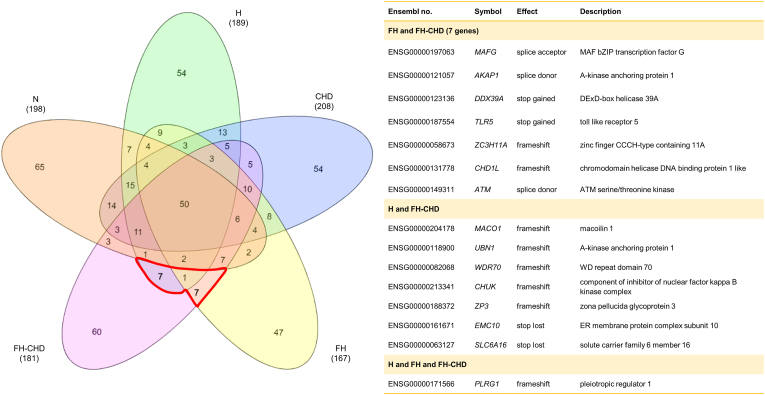 Figure 4
Figure 4Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsLipoproteins and Cardiovascular Health · Genetic Associations and Epidemiology · Diabetes, Cardiovascular Risks, and Lipoproteins
Introductions
1
Coronary heart disease (CHD) is a complex manifestation of atherosclerosis. The pathogenesis of the disease, which involves a long preclinical course, remains unclear. Various risk factors for CHD include dietary patterns, behaviors and lifestyles, for example, stress, smoking, high-fat food intake, decreased physical activity, inflammation, infectious diseases, abnormal endogenous blood constituents (such as lipids, lipoproteins, and coagulation proteins), adiposity, hypertension, and diabetes mellitus [1,2]. Moreover, genetic alterations are significantly associated with premature CHD.
Hyperlipidemia is an abnormal condition with high blood levels of lipoproteins or lipids, such as fats, triglycerides, cholesterol, and phospholipids, and is subdivided into two categories: primary (familial) and secondary (acquired) hyperlipidemia. Acquired hyperlipidemia is caused by other underlying disorders and lifestyles that cause alterations in plasma lipid and lipoprotein metabolism. In particular, hypercholesterolemia, characterized by high level of low-density lipoprotein (LDL) cholesterol, is one of the most prevalent risk factors in the progression of atherosclerosis and vascular disease involvement (e.g., CHD). Familial hypercholesterolemia (FH), a genetic disorder, is characterized by heritable and severely elevated levels of LDL-cholesterol [3] and mainly contributes to the development of premature cardiovascular disease (CVD), particularly premature or familial CHD (FH-CHD) [1,2]. Studies have reported that at least four genes related to sterol and lipoprotein pathways, including LDL receptors, apolipoprotein (apo) B, proprotein convertase subtilisin/kexin type 9, and the autosomal recessive hypercholesterolemia adaptor protein, are associated with cholesterol metabolism disorders [4,5]. Moreover, other roles of these proteins and associated receptors, or other genes linked to the FH-CHD development, should be investigated to evaluate the risk of developing this disease.
RNA-sequencing (RNA-seq) has been widely applied to detect gene expression in various tissues [6,7]. This technique also effectively detects transcriptome variants, such as single-nucleotide polymorphisms (SNPs) and short indels, in divergent tissues and species [[8], [9], [10], [11], [12]]. The present study aimed to explore intersected gene variants shared only between groups of hyperlipidemia, FH, and FH-CHD patients for their potential as interdependent predictors of the possibility of FH-CHD progression in hyperlipidemia patients. Next-generation sequencing (NGS)-based findings were used to determine if the selected variants had potential as markers for FH-CHD risk in patients with hyperlipidemia.
Material and methods
2
The corresponding author can provide all study materials, data, and analysis methods in this study upon reasonable request to repeat the results or simulate the procedures. In this cross-sectional study, male volunteers from Thailand, including healthy controls (N) and patients with hyperlipidemia (H), FH, CHD, and FH-CHD were enrolled. The RNA-seq or transcriptome data obtained from whole blood samples from each group (n = 3/group) were analyzed by NGS. The variant sequences found specifically in the H, FH, and FH-CHD groups were filtered to generate an intersecting gene variant profile. Using transcriptomics, we explored the genetic variants within genes that influence the development of CVD, as described previously [10].
Ethics approval and consent to participate
2.1
The present study was conducted at the Faculty of Tropical Medicine, Mahidol University, and approved by the Ethics Committee of Mahidol University (MUTM2017-025-01-2) and The Institutional Review Board, Royal Thai Army Medical Department (Q031b/59). Before recruitment, and after being informed of the study objectives, the volunteers completed informed consent forms.
Participants
2.2
Fifteen male volunteers born to Thai parents were recruited in the present study and assigned to the five groups (n = 3/group). Healthy volunteers without any infectious and underlying diseases or CVD risk factors were enrolled as controls (the N group). The remaining patients (n = 12), who were diagnosed and treated by a specialist (KP) at Phramongkutklao Hospital, were classified into the following groups based on the American College of Cardiology/American Heart Association criteria (2013) [13] patients with high cholesterol levels [total cholesterol (TC) > 200 mg/dL, LDL >130 mg/dL, and high-density lipoprotein (HDL) < 40 mg/dL] but with no evidence of vital organ dysfunction (H group); CHD patients scheduled to undergo coronary bypass grafting (CHD group); and three pairs of family related patients with FH and FH-CHD. The FH patients were identified according to clinical criteria (including TC > 240, LDL >170 with or without tendon xanthomas and corneal arcus) and familial history. All H, FH, CHD and FH-CHD patients were treated with appropriate cholesterol-lowering drugs since they were diagnosed as prescribed by KP. The workflow of this study is shown in Fig. 1. The clinical manifestations of all volunteers are described in Table 1.Fig. 1. Experimental design and study population.Fig. 1. Table 1Characteristics of the participants.Table 1. VariablesN *(n = 3)*Hyperlipidemia *(n = 3)*CHD *(n = 3)*FH *(n = 3)*FH−CHD *(n = 3)*Age (years)26 ± 5.1∗ (19−36)56.7 ± 4.4 (50−65)49.7 ± 4.5 (41−56)38.3 ± 9.3 (20−50)56.3 ± 11 (45−79)Lipid profileTriglycerides (mg/dL)108.3 ± 19.2 (83−146)163.3 ± 28.3 (134−220)245.7 ± 66.1 (141−368)165.0 ± 45.5 (117−256)19.0 ± 42.7 (125−271)Total cholesterol (mg/dL)∗∗174 ± 6.2 (165−186)192.3 ± 24.6 (148−233)160.3 ± 15.4 (131−183)233.7 ± 32.0 (171−276)193.0 ± 19.6 (171−232)HDL-cholesterol (mg/dL)48.3 ± 1.5 (46−51)58.7 ± 9.9 (45−78)44.7 ± 1.8 (42−48)57.0 ± 12.4 (35−78)45.3 ± 1.8 (42−48)LDL-cholesterol (mg/dL)104.0 ± 3.8 (98−111)101.0 ± 31.9 (43−110)66.3 ± 27.1 (15−107)144.0 ± 27.6 (89−175)109.7 ± 20.5 (80−149)**Complete Blood Count (CBC)**WBC (10^3^/μL)9.4 ± 2.1 (5.2−12.0)5.9 ± 0.3 (5.6−6.5)5.8 ± 0.2 (5.4−6.0)6.8 ± 0.9 (5.4−8.5)7.1 ± 0.9 (5.4−8.5)RBC (10^3^/μL)5.2 ± 0.4 (4.7−6.1)4.3 ± 0.4 (3.6−4.9)4.7 ± 0.1 (4.5−4.8)5.6 ± 0.4 (5.1−6.3)5.5 ± 0.7 (4.2−6.4)Hb (g/dL)14.8 ± 1.2 (13−17)13.5 ± 1.1 (11.2−14.7)13.7 ± 0.7 (12.5−15)14.6 ± 0.2 (14.2−15)14.3 ± 1.0 (13.3−16.2)HCT (%)43.2 ± 3.0 (38.6−48.8)39.3 ± 3.2 (32.9−43.3)40.1 ± 1.6 (38−43.1)43.2 ± 0.3 (42.6−46.8)43.3 ± 3.6 (38.3−49.7)LYMPH (%)37.1 ± 1.6 (35.4−40.2)30.1 ± 5.1 (21.4−39.1)32.8 ± 4.8 (23.3−38)34.5 ± 6.5 (25−47)30.1 ± 2.3 (27−34.5)MONO (%)4.9 ± 0.2 (4.6−5.2)6.0 ± 0.3 (5.3−6.4)6.1 ± 0.6 (5.3−7.3)4.6 ± 0.3 (4−5)6.6 ± 1.4 (4−8.7)All patients and controls were male. Data are shown as the mean ± SEM (min-max).N, normal controls; CHD, patients with coronary heart disease who were scheduled to undergo coronary bypass grafting; FH, patients with familial hypercholesterolemia; and FH-CHD, patients with familial coronary heart disease.HDL, high-density lipoprotein; LDL, low-density lipoprotein; WBC, white blood cells; RBC, red blood cells; Hb, hemoglobin; HCT, hematocrit; LYMPH, lymphocyte; MONO, monocyte.∗Age in N was significantly younger than H (p = 0.0106) and CHD groups (p = 0.0255),while not significantly different from FH and FH-CHD groups (p > 0.05).∗∗ LDL levels among all patient groups were not significantly different comparing to N group (all p > 0.05).
Blood sample collection and experimental design
2.3
Blood samples treated with heparin (5 mL) were collected from the all patients before hyperlipidemia treatment or coronary bypass grafting, as well as from healthy controls. Sera separated from 1 mL of clotted blood was used to detect lipid markers, including triglycerides (TG), TC, LDL, and HDL. Kits (Randox Laboratories Ltd., Crumlin, UK) and a biochemistry analyzer (Architect CI 16200, Abbott Laboratories, Abbott Park, IL, USA) were used to analyze the lipids enzymatically [14]. A QIAamp RNA Blood Mini kit (Qiagen Inc., Germantown, MD, USA) was used to immediately extract total RNA from approximately 2 mL of whole peripheral blood, which was then stored at −70 °C. High-throughput transcriptome sequencing was achieved by performing NGS, and the whole RNA-seq data were evaluated and mapped among the groups. Genetic variations found only within the coding regions of genes intersecting between the H/FH and FH-CHD groups were selected. The properties and functions of the selected variants were evaluated to find possible links with FH-CHD development.
cDNA library construction and NGS sequencing
2.4
Total RNA was collected from 15 samples (3 samples from each group). The amount and quality of all the total RNA samples were analyzed, and integrity was assessed using an Agilent 2100 Bioanalyzer. Only samples with a high RNA Integrity Number (RIN ≥8.0) were then subjected to cDNA library construction and NGS sequencing in duplicate, as described previously [14] at the Omics Sciences and Bioinformatics Center (Bangkok, Thailand). The statistical power of this experimental design, calculated in RNASegPower, was 0.53 [15].
Sequence alignment, variant identification, and annotation
2.5
Using bioinformatic analysis on whole RNA sequences, the variants were obtained, and their positions on genes were screened and filtered from all samples. Initial quality control of raw data files was performed using FASTQC software. Adapter sequence and low-quality reads were subsequently removed using Trimmomatic with the following parameters: ILLUMINACLIP: [AdapterFile]:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36 (specifying minimum base quality (Phred) of 3, minimum average quality of 15 over a 4-base window, and minimum read length of 36). The filtered reads were aligned to a human reference genome using the HISAT2 aligner. Variant calling was then conducted following the workflow outlined in Supplementary Method S1: Command Line Pipeline (Filtered_variants_called.vcf). Hard filtering criteria—such as “QD < 2.0” and “FS > 30.0”—were applied using GATK to obtain high-confidence variants [16]. The resulting variants were annotated with the Ensembl Variant Effect Predictor (VEP), which provided transcript-level consequences, affected genes, and functional classifications [17]. Additional biological annotation and relevant functional protein predictions were performed using NCBI (https://www.ncbi.nlm.nih.gov), GeneCards (https://www.genecards.org), and UniProt databases (https://www.uniprot.org).
Selection of gene variants focusing on coding regions share between H/FH and FH-CHD
2.6
The variant category counts and gene overlaps (intersected genes) were accessed from Venn diagram using InteractiVenn (web-based tool) [18]. Briefly, the VCF file (from 2.5) were annotated. Then only intersected gene variants sharing between H/FH, and FH-CHD groups focusing on high impact effect in coding region were selected. These variants were further determined properties, function and possibility associated with the progression of CHD based on reference databases.
Results
3
Characteristics of the patients and controls
3.1
The general descriptions and clinical manifestations of the H, FH, CHD and FH-CHD groups and the N group (n = 3/group) are shown as mean ± SEM (ranges, minimum-maximum values) in Table 1. The volunteers in the N group were significantly younger than those in the H (p = 0.0106) and CHD groups (p = 0.0255), while not significantly different from those in the FH and FH-CHD groups (p > 0.05). The age difference between the N group and the H and CHD groups is reasonable because the disease typically develops progressively with advancing age [1,2]. As all four patient groups received lipid-lowering medication, their LDL levels were not significantly different from those of the normal group (all p-values >0.05).
Preselection of the regulatory sequences
3.2
Several variants were screened from all samples in the N, H, FH, CHD, and FH-CHD groups by performing transcriptome sequencing (RNA-seq). The screening data revealed several variants in consequence (all) from all samples in each group. Focusing on the coding regions, the major variants included synonymous and missense variants. Minor variants included frameshift variants and in-frame deletions and insertions. The comparison between the number of significant gene variants in the four patient groups and the N group, the annotation analysis of all samples, and the severity of the impact are summarized in Fig. 2 and Table 2. Fig. 3 shows sample variants by groups in percentages mapped to biotypes, including protein coding, retained intron, non-sense-mediated decay, process transcript, IncRNA, and others.Fig. 2. Gene variation annotation of the whole genome performed by transcriptome sequencing (RNA-seq**)** analysis. Four patient groups, hyperlipidemia (H), familial hyperlipidemia (FH), coronary heart disease (CHD), and familial CHD (FH-CHD) compared with healthy volunteers (N). (A) Panels show variations in the whole genome (all consequences); (B) panels show variants in the coding sequence (coding consequences).Fig. 2. Table 2The counts of all variants of protein coding, and completely novel variants in each functional effect group separated by allele frequency category.Table 2. ConsequenceNHCHDFHFH−CHDImpact effectvariantsgenesvariantsgenesvariantsgenesvariantsgenesvariantsgenes3 prime UTR variantMODIFIER58211 (29725−111745)5356 (4943−5905)30230 (27682−34870)5246 (4719−5999)28762 (24822−34471)5104 (4117−5976)27451 (25775−28646)4569 (4236−4864)28234 (22061−32142)4530 (3720−5305)5 prime UTR variantMODIFIER8636 (5169−15269)1657 (1585−1738)4639 (3705−5318)1478 (1236−1702)4601 (3162−6103)1464 (1074−1912)4733 (4475−5040)1500 (1448−1574)4814 (3620−5711)1526 (1163−1794)coding sequence variantMODIFIER9 (5−15)6 (5−8)6 (4−7)6 (4−7)7 (3−−10)7 (3−10)9 (7−12)9 (7−12)9 (8−9)8 (8−9)downstream gene variantMODIFIER84069 (62783−126144)6428 (6034−6929)64652 (62059−69126)6349 (5923−7077)63203 (59837−67943)6203 (5317−6912)63411 (62725−63800)5666 (5351−5929)63468 (59047−66523)5892 (4897−6467)**frameshift variant^4^HIGH171 (98−276)****38 (33−44)****103 (77−123)****33 (27−40)****89 (71−112)****33 (31−36)****79 (58−109)****28 (22−37)****97 (86−116)****38 (30−46)**In-frame deletion variantMODERATE207 (87−447)28 (24−36)95 (81−117)24 (23−26)85 (64−108)23 (17−29)87 (72−108)24 (22−27)79 (46−105)24 (16−28)In-frame insertion variantMODERATE151 (51−313)18 (11−22)61 (55−64)15 (12−17)63 (43−83)14 (10−19)68 (57−77)16 (14−18)63 (55−72)15 (13−17)intron variantMODIFIER164247 (88896−294635)6041 (5853−6199)109962 (96910−120324)5959 (5741−6308)113890 (95446−125819)5984 (5270−6565)114298 (111866−116473)5511 (5273−5710)112980 (100608−131380)5752 (5101−6122)missense variantMODERATE16736 (10013−29715)2374 (2248−2501)9161 (8238−10306)2196 (1972−2481)8782 (7008−11021)2111 (1783−2612)8990 (8369−9657)13699 (1948−36950)9133 (6827−10453)2116 (1629−2482)**splice-acceptor variant^1^HIGH37 (15−55)****12 (11−13)****28 (24−31)****15 (13−18)****32 (20−−43)****17 (12−19)****41 (23−58)****18 (14−21)****26 (25−27)****15 (14−15)****splice donor variant^2^HIGH53 (35−79)****14 (11−13)****22 (17−25)****12 (9−17)****24 (18−36)****13 (8−19)****20 (9−31)****11 (7−15)****25 (23−26)****12 (11−14)**splice region variantLOW990 (618−1656)225 (209−237)628 (540−712)214 (180−244)622 (468−701)206 (170−226)628 (559−683)218 (212−229)2586 (659−6363)232 (226−241)**start-lost variant^6^HIGH31 (17−53)****11 (10−12)****22 (17−25)****13 (10−16)****20 (11−29)****13 (8−17)****17 (10−26)****9 (6−12)****34 (7−74)****10 (5−14)****stop-gained variant^3^HIGH44 (18−77)****14 (12−15)****17 (10−22)****11 (7−14)****23 (15−34)****12 (10−15)****19 (17−20)****12 (11−14)****20 (6−32)****10 (6−13)****stop-lost variant^5^HIGH14 (9−20)****8 (7−10)****11 (9−12)****9 (7−10)****11 (5−15)****9 (5−11)****8 (7−9)****7 (7−7)****9 (6−12)****9 (6−13)**stop-retained variantLOW14 (7−20)6 (5−8)10 (5−16)5 (4−7)11 (9−−15)6 (5−8)9 (5−12)6 (3−8)13 (8−22)7 (5−11)synonymous variantLOW23129 (13736−40435)2982 (2790−3257)13116 (11697−14860)2813 (2511−3210)12370 (10054−15625)2660 (2275−3317)12717 (11811−13463)2631 (2471−2724)12671 (9998−14234)2680 (2044−3063)upstream gene variantMODIFIER37820 (25267−60500)3730 (3654−3833)27184 (26030−28293)3603 (3374−3869)27659 (25720−29509)3618 (3354−3970)28292 (27391−29657)3502 (3449−3537)29047 (28144−30060)3561 (3128−3907All data are shown as the mean (min-max).∗1–6 are shown in order of severity (more to less severe) estimated by Ensembl as high impact.Fig. 3. Sample variants by group in percentage mapped to biotype.Fig. 3
Selection of intersecting target genes
3.3
The variants within the coding regions of the genes in all groups were assessed as illustrated in the Venn diagram (Fig. 4). We selected 15 intersecting variant genes with a high impact: MAFG, AKAP1, DDX39A, TLR5, ZC3H11A, CHD1L, ATM, MACO1, UBN1, WDR70, CHUK, ZP3, EMC10, SLC6A16, and PLRG1, which were shared between the H/FH and FH-CHD groups (Fig. 4). Functional analysis revealed that only 6 of 15 variant genes—CHUK, EMC10, PLRG1, MAFG, AKAP1, and TLR5—are potentially related to atherogenesis. The gene description, location, and effect of the variant and remarkable changes in the variant genes are shown in Table 3.Fig. 4. Venn diagram illustrates the variants focused on coding regions in the N, H, FH, CHD, and FH-CHD groups using transcriptome sequencing (RNA-Seq) analysis.The variant found in fifteen genes intersected between FH and FH-CHD (7 genes including MAFG, AKAP1, DDX39A, TLR5, ZC3H11A, CHD1L, and ATM), H and FH-CHD (7 genes including MACO1, UBN1, WDR70, CHUK, ZP3, EMC10 and SLC6A16), H and FH and FH-CHD (1 gene is PLRG1) were selected for determining as feasible associate for FH-CHD risk.Fig. 4. Table 3High-impact variants in the coding regions of seven variant gene intersections between the FH and FH-CHD groups.Table 3. Ensembl no. (Transcript ID)NCBI RefSeqSymbolLocation (Genomic)EffectsAlleles (Ref > Alt)Protein ChangeHGVS [19,20](Genomic) g.∗HGVS [19,20] (Protein) p.∗Pathogenicity Scoring (ACMG Criteria) [21]FH and FH-CHD: seven genesENSG00000197063 (ENST00000357736.9)NM_002359.4MAFG1365407:17:00splice acceptorC > G–chr17:g.81923417C > Gp.(?)
- ·Predicted loss-of-function (splice-site)
- ·VUS ENSG00000121057 (ENST00000337714.8)NM_003488.4AKAP1952021:55:00splice donorT > C–chr17:g.57120295T > Cp.(?)
- ·Predicted loss-of-function (splice-site);
- ·VUS ENSG00000123136 (ENST00000242776.9)NM_005804.4DDX39A240184:27:00stop-gainedG > AArg263Terchr19:g.14409927G > Ap.(Arg263Ter)
- ·Predicted loss-of-function (nonsense);
- ·VUS ENSG00000187554 (ENST00000642603.2)NM_003268.6TLR53718531:58:00stop-gainedG > AArg392Terchr1:g.223111858G > Ap.(Arg392Ter)
- ·Predicted loss-of-function (nonsense);
- ·VUS ENSG00000058673 (ENST00000367210.3)NM_001376342.1ZC3H11A3397457:48:00frameshiftIns AGln423fschr1:g.203847408insAp.(Gln423fs∗?)
- ·Predicted loss-of-function (frameshift);
- ·VUS ENSG00000131778 (ENST00000369258.8)NM_004284.6CHD1L1:147264583–147264584frameshiftDel GAla84fschr1:g.147264584delGp.(Ala84fs∗?)
- ·Predicted loss-of-function (frameshift);
- ·VUS ENSG00000149311 (ENST00000675843.1)NM_000051.4ATM11:108330421–108330424splice donorDel GCA–chr11:g.108330421_108330424delGCAp.(?)
- ·Predicted loss-of-function (splice-site);
- ·VUS H and FH-CHD: seven genesENSG00000204178 (ENST00000374343.5)NM_018202.6MACO1424149:03:00frameshiftIns TLeu100fschr1:g.25448883insTp.(Leu100fs∗?)
- ·Predicted loss-of-function (frameshift);
- ·VUS ENSG00000118900 (ENST00000262376.11)NM_001079514.3UBN181260:26:00frameshiftIns A-/Xchr16:g.4874666insAp.?
- ·Predicted loss-of-function (frameshift);
- ·VUS ENSG00000082068 (ENST00000265107.9)NM_018034.4WDR70626757:16:00frameshiftIns A-/Xchr5:g.37605136insAp.?
- ·Predicted loss-of-function (frameshift);
- ·VUS ENSG00000213341 (ENST00000370397.8)NM_001278.5CHUK1670086:03:00frameshiftIns A-/Xchr10:g.100204563insAp.?
- ·Predicted loss-of-function (frameshift);
- ·VUS ENSG00000188372 (ENST00000394857.8)NM_001110354.2ZP31274038:06:00frameshiftIns GVal186fschr7:g.76441866insGp.(Val186fs∗?)
- ·Predicted loss-of-function (frameshift);
- ·VUS ENSG00000161671 (ENST00000334976.11)NM_206538.4EMC10841397:48:00stop-lostG > T∗372Tyrchr19:g.50482728G > Tp.(Ter372Tyr)
- ·Predicted altered stop codon (stop-lost);
- ·VUS ENSG00000063127 (ENST00000335875.9)NM_014037.3SLC6A16821787:29:00stop-lostT > C∗34Trpchr19:g.49306109T > Cp.(Ter34Trp)
- ·Predicted altered stop codon (stop-lost);
- ·VUS H and FH and FH-CHD: one geneENSG00000171566 (ENST00000499023.7)NM_002669.4PLRG12575656:16:00frameshiftIns T-/Xchr4:g.154539136insTp.?
- ·Predicted loss-of-function (frameshift);
- ·VUS Ins; insert, Del; delete, fs; frameshift, Ter = “termination codon” = stop codon (UAA/UAG/UGA), p.(?) = Uncertain predicted protein effect, p.? = Unknown protein effect, ∗ = stop codon.Pathogenicity Scoring; Prediction = no segregation, function, population frequency, and several genes are still candidate genes for FH/H/FH-CHD, Variant of Uncertain Significance (VUS) = Insufficient information to conclude whether the variant causes disease [21].
Frameshift variations were found in eight genes—ZC3H11A, CHD1L, MACO1, UBN1, WDR70, CHUK, ZP3, and PLRG1—which disrupted the reading frame, probably resulting in a dysfunctional protein. Stop-gain variations were observed in DDX39A at the 6th exon and in TLR5 at the last intron, which added a premature stop codon, leading to a shortened transcript at amino acid positions 263 and 392, respectively. Stop-lost variations that alter the termination codon and lead to elongated proteins were noted in EMC10 and SLC6A16. MAFG harbored a splice-acceptor variant, whereas AKAP1 and ATM harbored splice donor variants.
The descriptions and roles of the six selected variants—CHUK, EMC10, PLRG1, MAFG, AKAP1, and TLR5—in the context of CVD and other vascular diseases, as reported in previous studies, are described in the Discussion section.
Discussion
4
Our recent previous study reported that the expressions of TRPM2, PDLIM5, BCL3, and GBA genes in patients with FH can potentially be used as predictive markers of FH-CHD [14]. These genes are involved in chronic inflammation and pathways involved in the CHD pathogenesis. In the present study, using the same volunteers as Prasongsukarn K et al., 2021(14), we attempted to use the alternative advantage of RNA-seq data to identify the potential gene variants in four patient groups and a control group to identify additional probable novel markers. Our findings revealed that the hyperlipidemia, FH, and FH-CHD groups shared several variants among six genes—MAFG, AKAP1, TLR5, CHUK, EMC10, and PLRG1—which are potentially associated with the progression of CHD. Several previous studies on patients with FH revealed that variants in genes coding for LDL-c receptors and lipid metabolic pathways, such as APOB, LDLR, PCSK9, or LDLRAP1, are important risk factors for the diagnosis of FH-CHD [[22], [23], [24], [25]].
However, the gene variants found in our study were not directly associated with major lipid-metabolism pathways. Our findings revealed genes involved in other pathways related to the complex atherogenic process and CHD development, which adds to previous knowledge [4,5,[22], [23], [24], [25]]. We emphasize that these findings are likely to support further in-depth study aimed at clarifying the potential of these variants for use as interdependent predictors of the possibility of FH-CHD progression in hyperlipidemia patients.
RNA-seq analysis has been widely applied to identify and assess the expression of genes in various tissues [6]. Furthermore, this technique also effectively detects transcriptome variants, such as SNPs and short indels, in divergent tissues and species [[8], [9], [10], [11], [12],26]. However, in present study, we applied only RNA-seq data to explore the gene variants in the coding regions. We did not screen exon DNA and short neighboring intron sequences as templates to validate the gene variants by Sanger sequencing or gDNA targeted NGS [27]. However, recent advances in NGS have enabled accurate identification of diverse genomic variants, which revolutionizes methods to obtain genomic data [22,24,26,28,29]. Using gene panels and exome and genome sequencing, diverse coding variants can be identified for several disorders [26,30].
In this preliminary study, based on a reference database without any gene variant validation, we attempted to link specific high-impact variants within selected genes—MAFG, AKAP1, TLR5, CHUK, EMC10, and PLRG1 – to their possible roles in atherogenesis and CVD.
MAFG encodes for a newly discovered key transcription factor involved in preventing apoptosis of vascular endothelial cells, which is the leading risk factor for atherosclerosis [31]. It also acts as a transcriptional repressor that modulates bile acid and cholesterol metabolism [32]. A previous in vitro study on apoptosis of human umbilical vein endothelial cells induced by palmitic acid revealed that MAFG and MAFF expression could inhibit apoptosis in these cells [31]. The MAFG protein acts synergistically with nuclear factor erythroid 2-like 2 (NFE2L2 or Nrf2) to transcriptionally repress or activate gene functions related to the antioxidant response element (ARE)-dependent pathways in skeletal muscle cells [33]. MAFF, MAFG, and NFE2L1 can transcriptionally activate ARE-associated genes cooperatively in arterial endothelial cells, which enhances the antioxidant levels in these endothelial cells, mitigating cell injury and suppressing atherogenesis [31]. Although a splice-acceptor variant was discovered in MAFG (Fig. 3, Table 2), no significant changes in the codon or amino acid position were observed. Splice-site variants are alterations of the DNA sequence occurring at the border of an exon and an intron (splice-site), potentially disrupting RNA splicing, resulting in the loss of exons or the inclusion of introns, and altering the protein-coding sequence [29,34].
AKAP1 encodes a protein belonging to the AKAP family, which binds to the types I and II regulatory subunits of protein kinase A and anchors them to the mitochondrion. Intact mitochondrial homeostasis is pivotal for effective contractile function and metabolism of cardiac muscles. Impaired mitochondrial dynamics contribute to the pathophysiology underlying various CVDs [35]. AKAP1 is potentially related to the cAMP-dependent signal transduction pathway and in regulating RNA to a specific cellular compartment. Therefore, this protein is associated with CVD, mitochondrial function, brain ischemia, hypoxia, EC, HT, lipid metabolism, and myocardial hypertrophy. Deletion of AKAP1 results in complex II dysfunction. Mitochondrial respiration, or the electron transport chain (ETC), is the main source of superoxide during ischemic and excitotoxic injuries. Therefore, variants in the subunits or assembly factors of the ETC can cause neurodegenerative disorders [36]. In this study, we noted a single splice donor variant in AKAP1. Similar to the MAFG variant, we found no significant changes in the codon and amino acid positions (Table 2).
TLR5 encodes a toll-like receptor (TLR) family member, which is crucial for pathogen recognition and activation of innate immunity through the detection of specific pathogen-associated molecular patterns. This gene encodes proteins that recognize bacterial flagellin, the main component of bacterial flagella and a virulence factor. The induction of this receptor triggers the activity of NF-κB, which in turn induces several target genes related to inflammation. The NF-κB family of transcription factors is an important regulator of immune development, immune responses, and inflammation. False regulation of NF-κB is implicated in numerous diseases ranging from inflammatory to immune disorders [37]. Several TLR subfamilies, including TLR2, TLR4, and TLR5, are linked to the initiation and progression of atherosclerotic lesions. In this study, a stop codon variant was noted in TLR5 that led to translation of a shortened loss-of-function TLR5 protein. This might affect various pathways related to NF-κB activation, affect immune responses to inflammation, and moderate atherogenesis process, as previously reported [37]. In addition, hematopoietic TLR5 deficiency prevents atherosclerotic plaque formation by reducing macrophage accumulation and impairing T-cell responsiveness [38].
CHUK is a protein-coding gene that encodes a member of the serine/threonine protein kinase family. The encoded protein is a unit of a cytokine-activated protein complex. In addition, this protein inhibits the fundamental transcription factor NF-κB complex, by phosphorylating sites that induce the inhibitor degradation via the ubiquitination pathway, thereby triggering the transcription factor [provided by RefSeq, Jul 2008]. CHUK is one of the detected fatty acid-associated genes, including genes involved in the metabolisms of lipid and/or carbohydrate, or in the arrangement of intramuscular fatty acid accumulation (e.g., ADIPOQ, CHUK, CYCS, CYP4B1, DLD, ELOVL6, FBP1, G0S2, GCLC, HMGCR, IDH3A, LEP, LGALS12, LPIN1, PLIN1, PNPLA8, PPP1R1B, SDR16C5, SFRP5, SOD3, SNW1, and TFRC) in pigs [39]. Moreover, CHUK plays a pivotal role in hypertension, and hyperlipidemia, which are common risk factors of ischemic stroke (IS). CHUK variants rs2230804 and rs3808917 may be associated with elevated blood pressure and lipid levels of IS patients [40].
EMC10 is a protein-coding gene that encodes 1 of 10 units of the endoplasmic reticulum complex (EMC), which is a highly conserved protein complex associated with the biology of membrane protein (summary by Ref. [41]). EMC10 has several roles, including contributing to membrane insertase activity, positively regulating angiogenesis, promoting endothelial cell proliferation, and facilitating protein insertion into the endoplasmic reticulum (ER) membrane. EMC10 is located in the endoplasmic reticulum membrane and the extracellular region [42]. Furthermore, secreted EMC10 (scEMC10) is upregulated in people with obesity and is correlated with insulin resistance. Overexpressed scEMC10 decreases energy consumption, thus promoting obesity in mice. scEMC10 is a circulating inhibitor of thermogenesis and a potential therapeutic target for obesity and its cardiometabolic complications [43]. EMC10 is produced by monocytes and macrophages derived from bone marrow. It functions as an angiogenic growth factor involved in myocardial infarction repair [44].
PLRG1 is a protein-coding gene that encodes a core unit of the cell-division cycle 5-like (CDC5L) complex Pleiotropic Regulator 1. The CDC5L complex is part of the spliceosome and is necessary for pre-mRNA splicing. The encoded protein plays a crucial role in alternative splice-site selection. Alternatively, spliced transcript variants encoding multiple isoforms have been observed for this gene (provided by RefSeq, Jan 2011). The PLRG1 variant reportedly is related to severe congenital human cardiovascular malformations [45]. In a mouse model, PLRG1 has an essential role as a critical nuclear regulator of p53-dependent cell-cycle progression and apoptosis during both embryonic development and adult tissue homeostasis [46]. Taken together, our findings revealed that the six target gene variants are potentially related to the risk of FH-CHD in patients with hyperlipidemia.
However, this study had some limitations. In this preliminary study, we conducted an exploratory analysis of gene variants with low statistical power (0.53), due to the small genetic sample size and single-center analysis. Further multi-center studies with larger sample sizes and gene variant validation, using either Sanger PCR or gDNA targeted NGS, should be conducted to evaluate the feasibility of key variants for assessing FH-CHD risk.
In addition, the findings of high-impact variations in the six genes and their effects were estimated on the basis of bioinformatic analyses. Further studies involving bioinformatic analyses, variant classification, and functional validation assays are required. This knowledge would help researchers understand the gene function, biological mechanisms of the predicted genes, and variant pathogenicity to identify better predictive, diagnostic, and prognostic methods for premature CHD [34,37].
Conclusions
5
Our preliminary study suggested that the variations in the MAFG, AKAP1, TLR5, CHUK, EMC10, and PLRG1 genes are related to the risk of premature CHD. Further classification and functional validation should be performed to confirm our results and support the use of these gene variants as indicative markers for FH-CHD risk in hyperlipidemia patients.
CRediT authorship contribution statement
Wilanee Dechkhajorn: Data curation, Formal analysis, Investigation, Methodology, Project administration, Validation, Visualization. Kriengchai Prasongsukarn: Conceptualization, Data curation, Methodology. Surachet Benjathummarak: Formal analysis, Investigation, Methodology, Validation. Supachai Topanurak: Supervision, Validation, Visualization. Yaowapa Maneerat: Conceptualization, Data curation, Funding acquisition, Methodology, Resources, Supervision, Visualization, Writing – original draft, Writing – review & editing.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Gonzalez-Jaramillo N.Wilhelm M.Arango-Rivas A.M.Gonzalez-Jaramillo V.Mesa-Vieira C.Minder B.Systematic review of physical activity trajectories and mortality in patients with coronary artery disease J. Am. Coll. Cardiol.791720221690170010.1016/j.jacc.2022.02.03635483757 · doi ↗ · pubmed ↗
- 2Sucato V.Coppola G.Manno G.Vadala G.Novo G.Corrado E.Coronary artery disease in South Asian patients: cardiovascular risk factors, pathogenesis and treatments Curr. Probl. Cardiol.488202310122810.1016/j.cpcardiol.2022.10122835500733 · doi ↗ · pubmed ↗
- 3Weber C.Noels H.Atherosclerosis: current pathogenesis and therapeutic options Nat. Med.171120111410142210.1038/nm.253822064431 · doi ↗ · pubmed ↗
- 4Defesche J.World health organisation report on familial hypercholesterolemia Atherosclerosis 1541200124210.1016/s 0021-9150(00)00646-811137105 · doi ↗ · pubmed ↗
- 5Marais A.D.Familial hypercholesterolaemia Clin. Biochem. Rev.2512004496818516203 PMC 1853359 · pubmed ↗
- 6Wang Z.Gerstein M.Snyder M.RNA-Seq: a revolutionary tool for transcriptomics Nat. Rev. Genet.1012009576310.1038/nrg 248419015660 PMC 2949280 · doi ↗ · pubmed ↗
- 7Xiao Y.Yang J.Fu Z.Xiong Z.Zhang C.He D.Inhibition of Galectin-3 impairs antifungal immune response in Fungal Keratitis Dis. Markers 20222022831600410.1155/2022/8316004 PMC 901328935437453 · doi ↗ · pubmed ↗
- 8Baker E.K.Han J.Langley W.A.Reott M.A.Jr.Hallinan B.E.Hopkin R.J.RNA sequencing reveals a complete picture of a homozygous missense variant in a patient with VPS 13D movement disorder: a case report and review of the literature Mol. Genet. Genom.298520231185119910.1007/s 00438-023-02044-y 37340120 · doi ↗ · pubmed ↗