Assessing the effect of bovine MSTN variants on pre‐mRNA splicing
Nicolas Gaiani, Dominique Rocha, Arnaud Boulling

TL;DR
This study evaluates how specific MSTN gene variants affect RNA splicing in cattle, focusing on their role in double muscling.
Contribution
The study introduces a full-length gene assay to experimentally validate MSTN splicing variants and assesses the performance of two deep learning tools for splicing prediction.
Findings
None of the five missense MSTN variants tested had an effect on RNA splicing.
SpliceAI and Pangolin predictions were fully consistent with the experimental results from the FLGA.
The FLGA system proved effective for analyzing MSTN splicing variants in cattle.
Abstract
The myostatin protein is a potent negative regulator of skeletal muscle growth encoded by the MSTN gene. MSTN loss‐of‐function variants lead to a particular cattle phenotype characterized by an increase in skeletal muscle mass, known as “double muscling” or “double muscled”. However, most of the MSTN causal variants that have been linked to this phenotype lack experimental validation. This is the case, for example, for the five missense MSTN variants reported to be causal according to the Online Mendelian Inheritance in Animals. RNA splicing plays a major role in regulating gene expression; therefore, exploring the effects of variants on RNA splicing may provide relevant information on their functional impact. Here, we have set up a full‐length gene assay (FLGA) to functionally assess MSTN splicing variants, and we have used it to test the five missense variants plus a well‐described…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
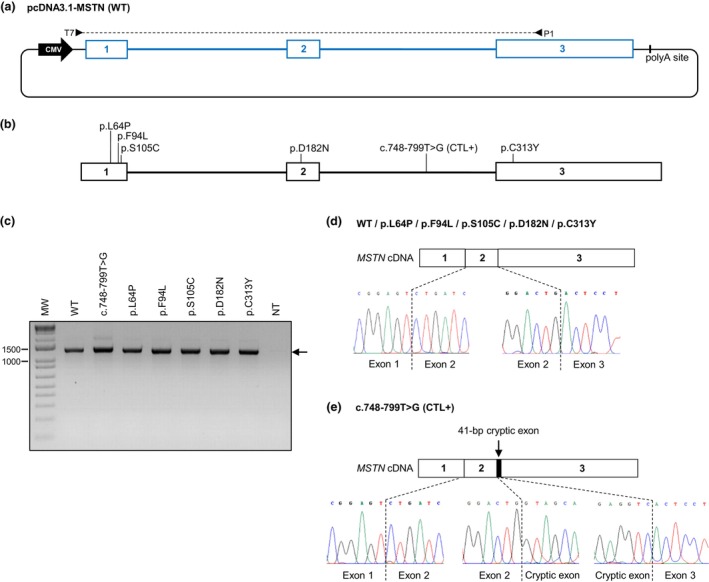 Figure 1
Figure 1- —Institut National de Recherche pour l'Agriculture, l'Alimentation et l'Environnement10.13039/501100022077
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMuscle Physiology and Disorders · RNA Research and Splicing · Genomics and Rare Diseases
The myostatin protein is a potent negative regulator of skeletal muscle growth encoded by the MSTN gene (Beyer et al., 2013). In cattle, only the canonical MSTN transcript has been identified, predominantly expressed in muscle, sperm, and uterus, with minimal or negligible expression observed in other tissues (Liu et al., 2022). Loss‐of‐function (LOF) variants in the MSTN gene are known to be responsible for an increase in skeletal muscle mass in different livestock species, including dog, sheep, cattle, and pig (Aiello et al., 2018). This characteristic muscle phenotype linked to MSTN alterations is monogenic and recessive and is referred to as “double muscling” or “double muscled” (DM). The first description of an LOF variant responsible for this trait in cattle was the c.821del11 mutation reported in Belgian Blue (Grobet et al., 1997). Several other LOF variants responsible for the DM phenotype have been identified later in additional breeds, including Blonde d'Aquitaine, Charolaise, Limousine, Marchigiana, and Piedmontese (Aiello et al., 2018; Kostusiak et al., 2023).
The functional interpretation of MSTN variants is straightforward when they cause a frameshift or truncation in the protein, thereby resulting in the loss of the essential C‐terminal active domain (McPherron et al., 1997; McPherron & Lee, 1997). However, the causality of a genetic variant in the elaboration of a given phenotype is not always obvious. For instance, the functional interpretation of missense variants is challenging. This class of variant may alter the function of proteins, as well as the amount and the sequence of RNA transcripts, but it may also be neutral. Typically, only the effects of amino acid substitutions are investigated to assess the variant's impact on protein function. However, splicing disruption caused by missense variants is relatively common and has been well documented in humans (Soemedi et al., 2017). This possibility should not be overlooked in farm species such as cattle. Understanding how variants impact splicing may help pinpoint causal variants. Identifying the causal variants involved in agronomic traits is important for understanding the underlying molecular mechanisms and also for developing more accurate genomic predictions through the integration of this information into the evaluation models (Liu et al., 2020).
Splicing is the process by which introns are removed from the primary RNA to obtain a mature RNA and it plays a major role in gene expression. We recently explored the role of splicing variants in the elaboration of cattle phenotypes using a massively parallel reporter assay (Charles et al., 2025). We also designed a full‐length gene assay (FLGA) to specifically assess the effect of DGAT1 variants on pre‐mRNA splicing (Gaiani et al., 2023). Using it, we confirmed the previously described effect of the p.M435L missense variant on the skipping of the DGAT1 exon 16 (Gaiani et al., 2023; Lehnert et al., 2015). Such assays can provide a better understanding of the functional impact of genetic variants, helping to classify them as causal or not.
Here, we designed an FLGA to analyze putative MSTN splicing variants. We used it to address the hypothesis that bovine MSTN missense variants responsible for the DM phenotype may decrease the function of the gene via splicing disruption. To retrieve such variants, we browsed the Online Mendelian Inheritance in Animals (OMIA) database (https://omia.org/home/) and found five missense variants assigned to the DM phenotype (OMIA:000683‐9913): p.L64P (Dierks et al., 2015), p.F94L (Grobet et al., 1998), p.S105C (Dunner et al., 2003), p.D182N (Dunner et al., 2003), and p.C313Y (Kambadur et al., 1997). The deep intronic c.748‐799T>G variant, which was described in vivo to create a cryptic exon, was included in our study to serve as a positive control (Bouyer et al., 2014). In addition, we used SpliceAI and Pangolin programs to predict the effect of these six variants on MSTN splicing, as we have recently validated them for use in cattle (Charles et al., 2025). This enabled us to assess whether these bioinformatic tools could be useful in classifying and filtering MSTN variants before any functional validation by FLGA. This can be relevant if a large number of variants need to be analyzed.
To implement the FLGA, the whole sequence of the bovine MSTN gene (ENSEMBL:ENSBTAG00000011808) has been introduced in the pcDNA3.1 expression vector using the In Fusion HD cloning Kit (Takara) to obtain the pcDNA3.1‐MSTN wild‐type (WT) construct (Figure 1a). A Methods section is provided in Appendix S1. The sequence inserted downstream of the CMV promoter began within MSTN Exon 1 and ended within Exon 3, spanning both translation start (ATG) and stop (TGA) codons (Figure 1a). Alternative alleles relative to the five missense variants and the deep intronic c.748‐799T>G variant were then introduced into the pcDNA3.1‐MSTN WT construct using the QuikChange II XL Site‐Directed Mutagenesis Kit (Agilent Technologies) to generate six different pcDNA3.1‐MSTN variant constructs (Figure 1b). pcDNA3.1‐MSTN WT and variant constructs were transfected in the HEK293T cells, which is a widely used model to perform splicing functional tests. The choice of this cell line was detailed previously (Charles et al., 2025). Forty‐eight hours after transfection, total RNA was extracted from transfected cells, and reverse transcription‐PCR were performed using PCR primers T7 and P1 to specifically amplify the cDNA originated from MSTN mRNA transcribed from the pcDNA3.1‐MSTN WT and variant constructs (Figure 1a). The resulting PCR products were analyzed by gel electrophoresis to check the size of the obtained amplicons. Finally, the same PCR products were purified on columns using the QIAquick PCR Purification Kit (Qiagen) and analyzed by Sanger sequencing.
Gel electrophoresis of the reverse transcription‐PCR products showed a single specific amplicon for the c.748‐799T>G variant, as well as for the five missense variants, of a size similar to those obtained with the WT construct (Figure 1c). Sanger sequencing of all of these purified PCR products revealed that the exact sequence of both MSTN introns has been fully spliced for each of the five missense variants, as well as for the WT (Figure 1d). This generated a 1317‐bp product that corresponds to the Ensembl canonical MSTN transcript (ENSEMBL:ENSBTAT00000015674), leading to the conclusions that: (i) the pcDNA3.1‐MSTN WT construct yielded the expected transcript; and (ii) these missense variants have no effect on splicing. By contrast, the unique mRNA yielded by the c.748‐799T>G variant construct corresponded to a transcript carrying a 41‐bp cryptic exon localized between exons 2 and 3 (Figure 1e). The sequence of this abnormal transcript was identical to that described in vivo by Bouyer et al. (2014) in homozygous carriers of the c.748‐799T>G variant in the Blonde d'Aquitaine breed. Of note, this was the most abundant MSTN transcript detected in homozygous c.748‐799T>G animals since the canonical form was barely detectable. This is concordant with the result of our test in which only the transcript that included the 41‐bp cryptic exon has been detected for this variant. If the normal transcript had also been generated, heterozygous positions would have been observed in the electropherogram, due to a mix of normal and abnormal transcripts (Figure 1e).
The five missense variants and the deep intronic c.748‐799T>G variant were analyzed using SpliceAI and Pangolin (Table 1). A score threshold of 0.2 is usually selected to predict splicing variants using both programs (Charles et al., 2025; Jaganathan et al., 2019; Zeng & Li, 2022). Prediction scores obtained for the five missense variants were 0 or 0.01 in any tested conditions, i.e. a gain or a loss of an acceptor or a donor splicing site. This means that according to SpliceAI and Pangolin, they are very unlikely to modify splicing. Scores obtained for the c.748‐799T>G splicing variant validated in vivo were largely above 0.2 with SpliceAI (acceptor gain: 0.73; donor gain: 0.66) and Pangolin (gain: 0.39). Moreover, it is worth noting that if we consider the positions of both new splicing sites predicted by SpliceAI, they were expected to lead to the inclusion of a cryptic exon which exactly corresponded to the 41‐bp cryptic exon observed in vivo and in our FLGA (Table 1).
Considering these findings, we can conclude that the five missense variants do not exert their functional effect through a splicing‐related mechanism or else in a minority way, which was not detected by our approach. To our knowledge, no evidence of abnormal splicing was previously reported to be associated with one or more of these variants in vivo, although the bovine muscle transcriptome has been analyzed by RNA‐seq (Guillocheau et al., 2019; Liu et al., 2022). However, abnormal transcripts may be missed in vivo depending on the breeds analyzed and the method used to map RNA‐seq data. To clarify this situation, we wanted to experimentally assess the splicing hypothesis for these five missense variants. Even in the case of negative results, this allows us to focus future functional investigations on other processes involved in gene expression. An impact at the level of the protein function represents a relevant hypothesis to explain why these missense variants are LOF, but no experimental validation has been carried out so far (Johnsson & Jungnickel, 2021). For example, p.L64P was predicted to be deleterious by PolyPhen and SIFT programs based on the physicochemical and evolutionary properties of amino acids (Dierks et al., 2015). Using an approach calculating the impact of missense polymorphisms on the structural and functional characteristics of proteins, the p.C313Y variant was predicted to alter MSTN protein structure and function, whereas the p.F94L variant was not found to clearly affect it (Peka & Balatsky, 2024).
Although we did not highlight any effect of MSTN missense variants on splicing, we still have produced and validated a biologically relevant system to functionally analyze variants of this gene. The use of our MSTN FLGA may be valuable in validating the causality of a splicing variant in vitro, especially when tissue samples of the carrier animal are not available. In silico screening using SpliceAI and Pangolin programs followed by FLGA represents an efficient tool to search for and validate MSTN splicing variants at a large scale. In addition, it enables the functional mechanisms of splicing variants to be explored in depth by testing particular environmental conditions (e.g., exposure to a compound such as cycloheximide) (Zou et al., 2016) or interactions with other variants that may be introduced into the plasmid constructs (Nielsen et al., 2007). Finally, the pcDNA3.1‐MSTN WT and variant constructs we generated may also be used to study the normal or mutated MSTN protein in a cell model since they yield a full transcript carrying the entire MSTN coding sequence. The plasmid constructs produced in this study have been deposited in Addgene (see Data Availability Statement).
AUTHOR CONTRIBUTIONS
A.B.: conceived and designed this study. N.G. and A.B.: managed this study. N.G.: performed laboratory experiments. N.G. and A.B.: analyzed the data. A.B.: writing—original draft preparation. N.G., D.R. and A.B.: writing—review and editing. All authors have read and agreed to the published version of the manuscript.
CONFLICT OF INTEREST STATEMENT
The authors declare no competing interests.
Supporting information
Appendix S1.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Aiello, D. , Patel, K. & Lasagna, E. (2018) The myostatin gene: an overview of mechanisms of action and its relevance to livestock animals. Animal Genetics, 49(6), 505–519.30125951 10.1111/age.12696 · doi ↗ · pubmed ↗
- 2Beyer, T.A. , Narimatsu, M. , Weiss, A. , David, L. & Wrana, J.L. (2013) The TGFβ superfamily in stem cell biology and early mammalian embryonic development. Biochimica et Biophysica Acta (BBA)—General Subjects, 1830(2), 2268–2279.22967760 10.1016/j.bbagen.2012.08.025 · doi ↗ · pubmed ↗
- 3Bouyer, C. , Forestier, L. , Renand, G. & Oulmouden, A. (2014) Deep intronic mutation and pseudo exon activation as a novel muscular hypertrophy modifier in cattle. P Lo S One, 9(5), e 97399.24827585 10.1371/journal.pone.0097399 PMC 4020855 · doi ↗ · pubmed ↗
- 4Charles, M. , Gaiani, N. , Sanchez, M.P. , Boussaha, M. , Hozé, C. , Boichard, D. et al. (2025) Functional impact of splicing variants in the elaboration of complex traits in cattle. Nature Communications, 16(1), 3893.10.1038/s 41467-025-58970-5PMC 1202228140274775 · doi ↗ · pubmed ↗
- 5Dierks, C. , Eder, J. , Glatzer, S. , Lehner, S. & Distl, O. (2015) A novel myostatin mutation in double‐muscled German Gelbvieh. Animal Genetics, 46(1), 91–92.25515003 10.1111/age.12242 · doi ↗ · pubmed ↗
- 6Dunner, S. , Miranda, M.E. , Amigues, Y. , Cañón, J. , Georges, M. , Hanset, R. et al. (2003) Haplotype diversity of the myostatin gene among beef cattle breeds. Genetics, Selection, Evolution, 35(1), 103–118.10.1186/1297-9686-35-1-103PMC 273268512605853 · doi ↗ · pubmed ↗
- 7Gaiani, N. , Bourgeois‐Brunel, L. , Rocha, D. & Boulling, A. (2023) Analysis of the impact of DGAT 1 p.M 435L and p.K 232A variants on pre‐m RNA splicing in a full‐length gene assay. Scientific Reports, 13(1), 8999.37268760 10.1038/s 41598-023-36142-z PMC 10238528 · doi ↗ · pubmed ↗
- 8Grobet, L. , Martin, L.J. , Poncelet, D. , Pirottin, D. , Brouwers, B. , Riquet, J. et al. (1997) A deletion in the bovine myostatin gene causes the double‐muscled phenotype in cattle. Nature Genetics, 17(1), 71–74.9288100 10.1038/ng 0997-71 · doi ↗ · pubmed ↗