Catellani-Inspired BN-Aromatic Expansion: A Versatile Tool toward π‑Extended 1,2-Azaborines with Tunable Photosensitizing Properties
Federica Rulli, Sergi Ordeix, Roger Bresolí-Obach, Santi Nonell, Josep Saurí, Cristina Ribas-Font, Alexandr Shafir, Raimon Puig de la Bellacasa, Ana B. Cuenca

TL;DR
This paper introduces a new method to create BN-containing aromatic compounds with tunable light-sensitizing properties for optically responsive materials.
Contribution
A versatile Catellani-inspired synthesis method for π-extended 1,2-azaborines with tunable photosensitizing properties is developed.
Findings
The method enables the synthesis of BN-embedded polyaromatic cores with diverse ring structures.
The 'boron-up' BN isostere shows over 10-fold higher singlet oxygen generation compared to the inverted isomer.
The approach allows direct comparison of the effect of BN orientation on photosensitizing properties.
Abstract
BN-isosterism, the replacement of carbon–carbon units with boron–nitrogen pairs in organic frameworks, offers a powerful means to create novel compounds, yet methods to access larger BN-containing polyaromatic cores remain scarce. Leveraging our recently developed multigram-scale synthesis of BN-naphthalene, we now combine it with a Catellani-type arene extension (Pd(OAc)2/P(2-furyl)3, norbornene) to rapidly access diverse extended BN-embedded polyaromatic cores. This strategy delivers BN-embedded benzo[c]phenanthridines and curved 8- and 7-membered ring-fused derivatives, as well as BN-embedded benzofluorenones in both normal and inverse BN-vector orientations. Importantly, the ability to access both directional BN isomers, in addition to the parent CC core, provides a rare opportunity to directly interrogate the effect of the presence and sense of the BN moiety. Most notably,…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 1
1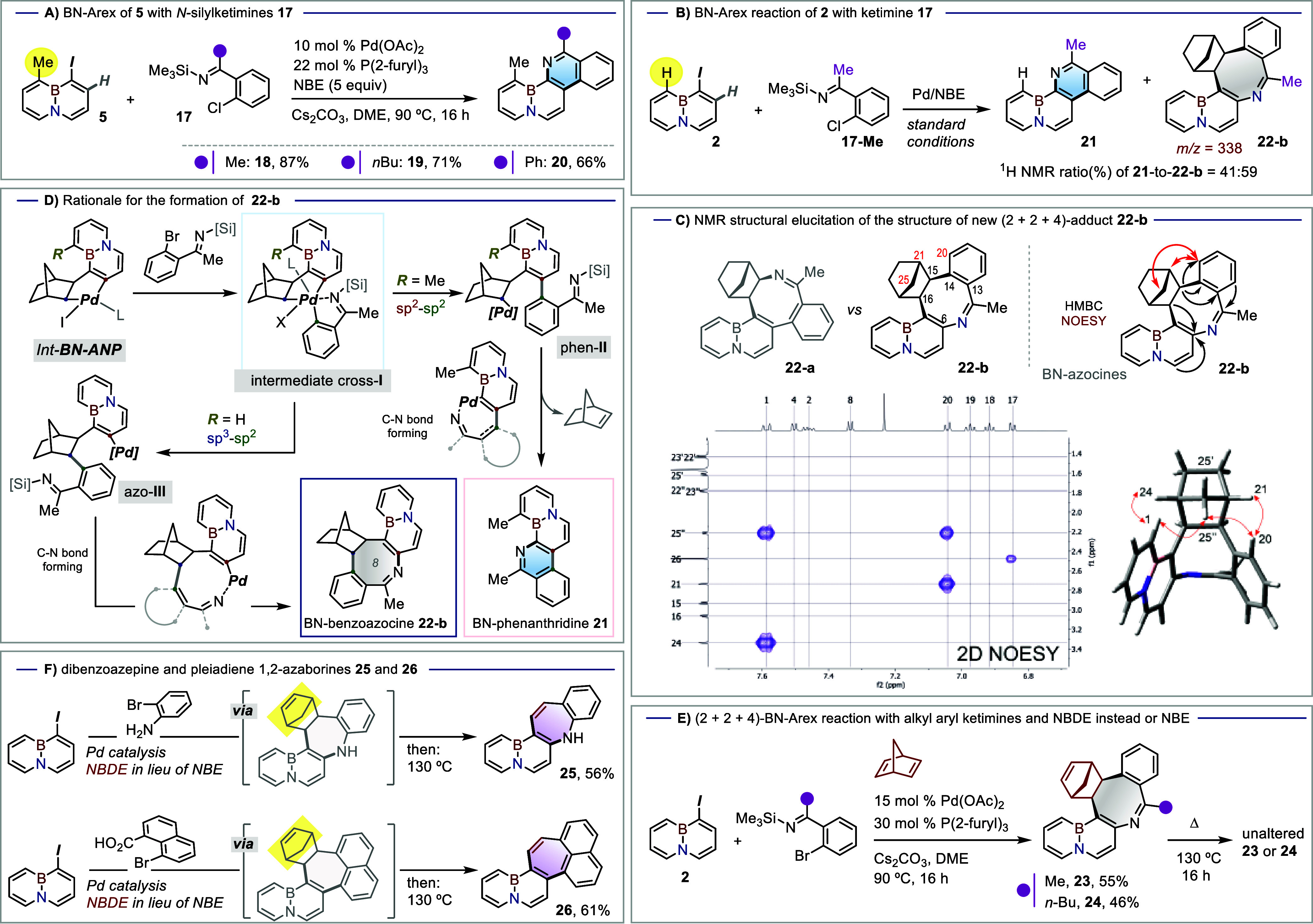 1
1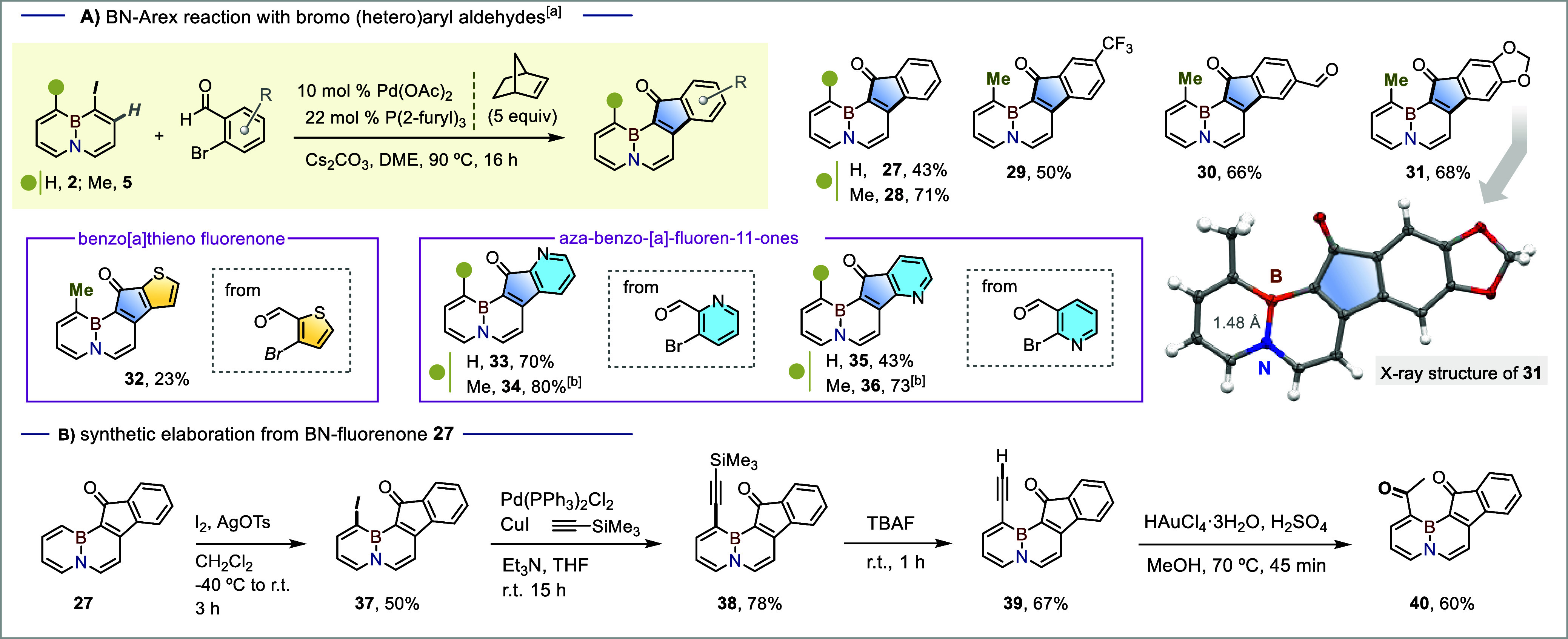 2
2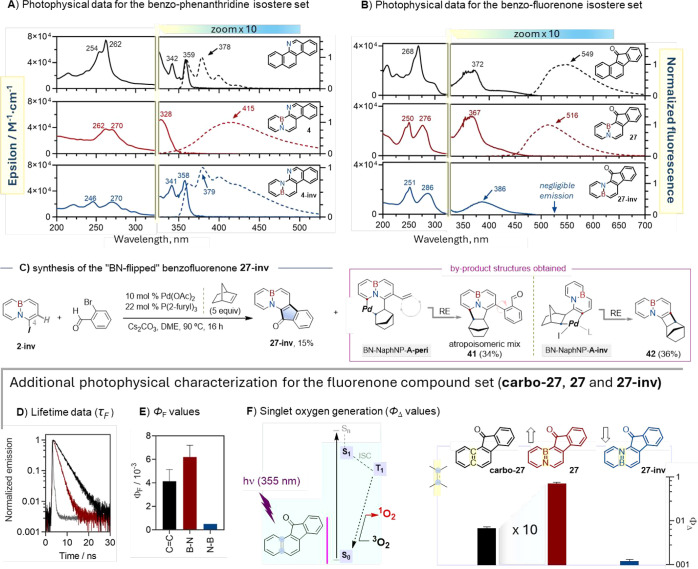 2
2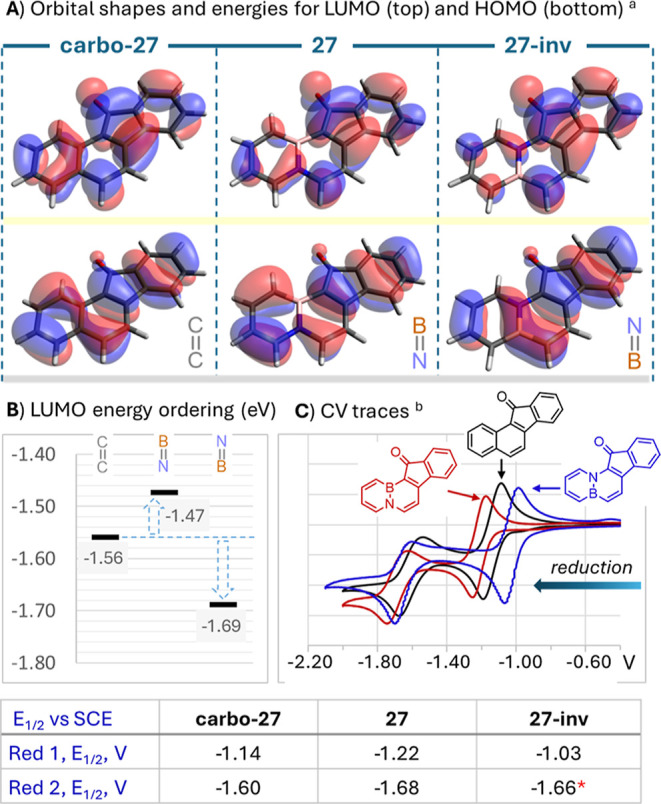 3
3- —Ministerio de Ciencia, Innovaci?n y Universidades10.13039/100014440
- —Ministerio de Ciencia, Innovaci?n y Universidades10.13039/100014440
- —Ministerio de Ciencia, Innovaci?n y Universidades10.13039/100014440
- —Ministerio de Ciencia, Innovaci?n y Universidades10.13039/100014440
- —Ministerio de Ciencia, Innovaci?n y Universidades10.13039/100014440
- —HORIZON EUROPE European Innovation Council10.13039/100018703
- —Ag?ncia de Gesti? d'Ajuts Universitaris i de Recerca10.13039/501100003030
- —Ag?ncia de Gesti? d'Ajuts Universitaris i de Recerca10.13039/501100003030
- —Instituci? Catalana de Recerca i Estudis Avan?ats10.13039/501100003741
- —European Regional Development Fund10.13039/501100008530
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsOrganoboron and organosilicon chemistry · Covalent Organic Framework Applications · Synthesis and Properties of Aromatic Compounds
Introduction
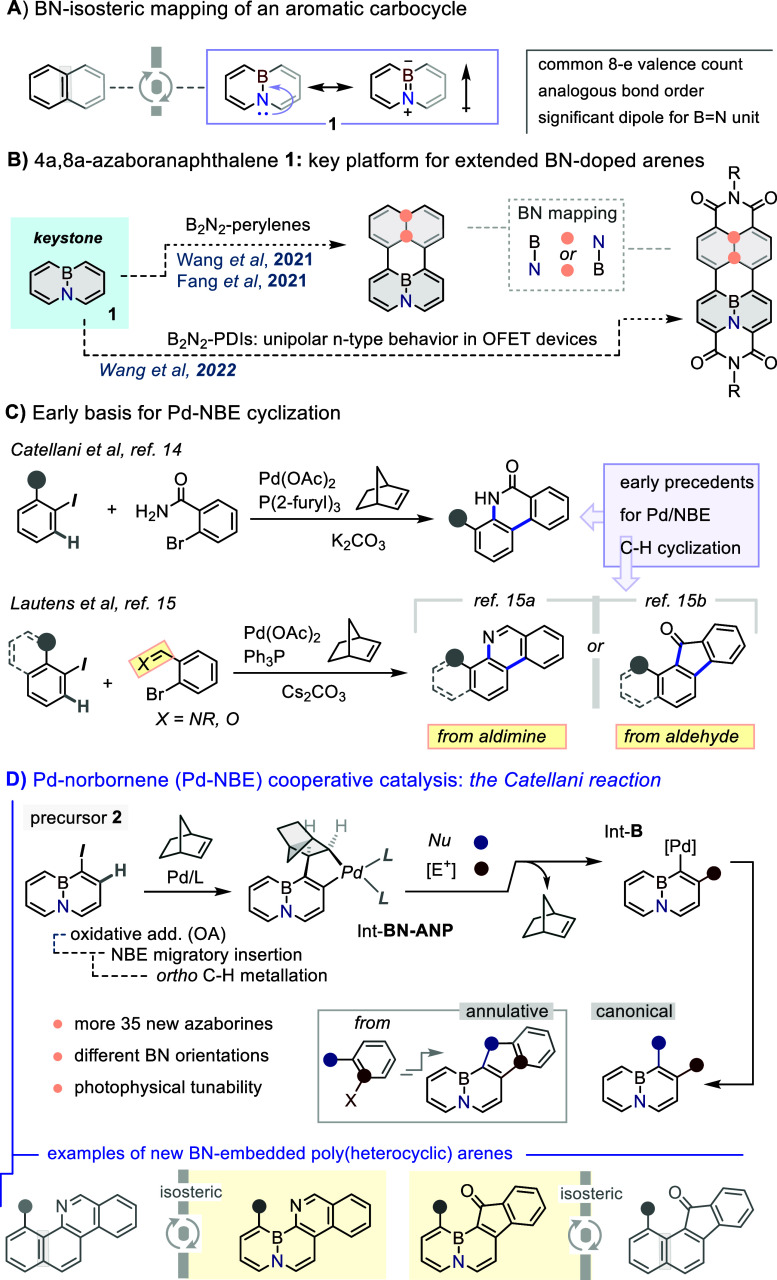
The replacement of one or more carbon–carbon fragments with isoelectronic boron–nitrogen pairs has emerged as a powerful strategy to modulate the electronic and photophysical properties of polycyclic aromatic hydrocarbons (PAHs).? While largely preserving the geometry of the parent core, BN-isosteric substitution introduces a highly polarized B–N unit (FigureA) that markedly alters the molecular orbitals. As a result, considerable efforts are directed toward incorporating 1,2-azaborine motifs into biomedical,? materials, ?,?,?,?,? and catalysis applications.? Progress, however, is hindered by the limited availability of systematic research on accessing BN-isosteres of even the smallest extended aromatic cores, particularly for targets in which the BN unit is embedded rather than located peripherally. Not surprisingly, this methodological scarcity is currently driving intensive efforts toward both the from-scratch construction and late-stage modification of azaborines. ?,?
(A) BN-isosterism concept. (B) BN-naphthalene 1 as gateway to larger aromatic BN systems. (C) Early precedent for Pd-NBE cyclizative difunctionalization. (D) General scheme and main steps of the Catellani-type Pd-NBE cycle, and examples of annulative ring extension of BN-naphthalene presented herein.
As part of our own efforts in this area, we recently described a unified strategy for accessing BN-embedded polycyclic cores enabled by the formation of aminoboranes through quantitative aminolysis of B(allyl)3 with N,N-diallyl and dipropargylamines.? While the strategy could potentially enable BN-mapping of larger PAH, such as anthracene and tetracene, it was also used in a robust multigram synthesis of the smaller 4a,8a-azaboranaphthalene 1, a BN-embedded isostere of naphthalene. This ready access to 1 might be expected to enable the synthesis of other BN-embedded PAH through downstream elaboration. Indeed, while various simpler functionalizations of 1 are known,? its use as a building block to form tri- and tetra-cyclic cores has remained scarce.? Nevertheless, the potential of 1 for accessing electronically versatile polyaromatic systems was recently illustrated in the synthesis of BN-doped perylenes,? perylenes-diimides? and terylene-diimides (FigureB).? The electronic tuning resulting from the isosteric replacement in these prototypical discrete organic semiconductors enabled, for example, the observation of a high unipolar n-type behavior through their inclusion in single-crystal OFETs.?
Seeking to further build on the naphthalene-like 1, we envisaged that metal-catalyzed annulative methodologies, such as a Catellani-type Pd/norbornene catalyzed annulative functionalization of 1, could potentially provide access to a large collection of new BN-doped PAHs, if the catalytic cycle proves compatible with the presence of a BN unit. This approach would employ an iodinated derivative of 1, such as 1-iodo-4a,8a-azaboranaphthalene 2, and would engage the C–H position ortho to iodine via norbornene (NBE)-promoted shuttling of the Pd center between adjacent carbons. ?,? Specifically, we were inspired by annulative variants of the Catellani reaction using tethered nucleophile–electrophile partners, an avenue elegantly pioneered by the Catellani? and Lautens laboratories? and subsequently extended by others.? Hence, Ferraccioli, Catellani and co-workers described the synthesis of phenanthridinones using 2-bromobenzamide as a dual coupling partner in combination with the Pd(OAc)2/tri(2-furyl)phosphine/NBE catalyst system (FigureC-top).? Furthermore, Lautens and co-workers demonstrated that employing 2-halo-aldimines and 2-halo-aldehydes as coupling partners with a related Pd/L/NBE system enables a modular synthesis of phenanthridine? and fluorenone cores (FigureC-bottom).?
Considering the far-reaching potential of this approach for BN-mapping key polyaromatic cores, the latter two manifolds were selected as starting point in the annulative ring expansion of BN-naphthalene 1. Hence, following an initial validation of the approach, this study integrates the multigram synthesis of 1 with a Pd/NBE-catalyzed arene expansion (ArEx) to deliver a range of 5, 6, 7 or 8-fused BN-π-extended polyarenes, notably benzo[c]phenanthridines and benzofluorenones. Among other advances, the method is shown to enable isosteric mapping in both orientations of the BN vector, thus allowing for systematic evaluation of the influence of this directional parameter on the electronic properties of the target cores. As an example, BN-mapping of the benzofluorenone core reveals a drastically enhanced singlet oxygen generation by the BN-embedded “boron-up”, but not by the “boron-down” isostere.
The mechanistic framework for our proposal is outlined in FigureD. Based on literature precedents, we envisage a sequence that begins with oxidative addition of the halo-derivative (e.g., 2) to a Pd(0) center, followed by olefin (NBE) insertion and ortho-C–H palladation via concerted metalation–deprotonation (CMD), yielding the key BN-naphthyl–norbornyl palladacycle Int-BN-ANP.? At this stage, incorporation of the electrophilic component at the ortho (C2) position occurs through oxidative addition of the E–Y reagent to give a Pd(IV) intermediate, followed by reductive elimination and NBE extrusion, which returns the metal center to the original C1 (ipso-Pd) position (intermediate B). The cycle is closed by reaction with Nu–X to regenerate Pd(0), ultimately leading to the vicinal incorporation of an electrophile and a nucleophile (FigureD).
Results and Discussion
First Results and Hypothesis Validation
Given the requirement for a halogenated BN-arene as entry point, we leveraged our earlier synthesis of the BN-naphthalene 1 ? to secure gram quantities of the iodo-azaborinine 2. The nonaromatic BN-bicycle 1-H _ 4 _ was procured through a one-pot process involving a solventless proto-deallylative condensation of triallylborane with N,N-diallylamine, followed by double ring-closing olefin metathesis (TableA). The subsequent aromatization to give 1 was achieved with Pd/C and norbornene as sacrificial H_2_ acceptor uniquely capable of preventing detrimental saturation of one of the two rings due to inefficient H_2_ removal.? While the iodination of 1 was previously achieved using the N-iodosuccinimide/AlCl_3_ combination,? in our hands this led to non-negligible amounts of the chlorinated side product, complicating product purification. Instead, the use of I_2_ activated with AgOTs provided 2 in a clean 85% yield (TableA).
1: Synthesis of 2 and Initial Shaping of the General Conditions for the (4 + 2)-BN-Arex Method
Inspired by the arene expansion precedent from the Lautens laboratory,? the formation of the BN-phenanthridine scaffold was attempted by exposing a mixture of 2 and 2-bromophenyl-N-silyl-aldimine, 3-Br, in acetonitrile to the combination of Pd(OAc)2/PPh_3_ with norbornene (NBE, 5 equiv) in the presence of Cs_2_CO_3_ (TableB). After 16 h at 90 °C, the GCMS analysis revealed the formation of a mixture of new species, including a peak consistent with the target BN-embedded phenanthridine 4 (m/z 230). Upon its isolation in 20% yield (TableB, entry 1), the structure of 4 was confirmed by ^1^H, ^13^C, ^11^B NMR spectroscopy (see Supporting Information); the yield was increased to 30% by switching to 1,2-dimethoxyethane (DME) as solvent (entry 2). While no significant improvement was gained by switching the ligand to tri(o-tolyl)phosphine or tricyclohexylphosphine (entries 3 and 4), the use of tri(2-furyl)phosphine resulted in a synthetically meaningful 50% yield (entry 5). The norbornene loading of 5 equiv proved optimal, with lower efficiencies observed when going below or above this amount (entries 6 and 7). This GCMS analysis also showed the reaction balance made up of BN-naphthalene side products containing a norbornyl fragment,? indicating hurdles in reaching or completing the norbornene extrusion stage. Considering that the well-documented ortho effect? might help alleviate some of these hurdles through additional steric bulk at the substrate’s complementary ortho position, we synthesized the iodo-BN naphthalene 5 bearing a peri-Me group (see Supporting Information for details). To our delight, the use of 5 under the previously optimized conditions led to the corresponding BN-phenanthridine 6 in 80% yield (TableB, entry 8); interestingly, the use of the chlorinated silyl imine 3-Cl proved similarly effective (77%, entry 9).
New BN-Phenanthridine Skeletons
The new (4 + 2)-BN-Arex variant was subsequently extended to other 2-bromoaryl-N-silyl-aldimines (Table). In this way, the extension of 2 with the CF_3_-substituted aldimine 7 afforded the BN-phenanthridine 8 in 53% yield; a more efficient reaction with the methylated precursor 5 afforded the peri-Me derivative 9 in 79%. The coupling between 5 and the heliotropin-derived aldimine afforded the pentacyclic BN-target 10 in a 47% yield. As an important synthetic feature, the process also provided rapid access to new family of poly heteroatom doped polycyclic cores. Hence, the use of the thiophene-based N-silyl aldimine 11 led to the smooth formation of the tetracyclic BN-benzo-thieno-[2,3-c]-quinolines 12 and 13 in 72% and 88% yields, respectively. Furthermore, the use of the nicotine-derived aldimine 14 led to the poly heterocyclic BN-naphthyridine cores 15 and 16 in 47% and 82% yield, respectively, albeit using an increased catalyst loading. Incidentally, while primarily targeting the poly heteroarenes 12–13 and 15–16 for their potential optoelectronic behavior, we note that both the thienoquinoline and naphthyridine units are also structural components of known pharmacophores, including those involved in kinase inhibition,? as well as anticancer and antiviral scaffolds.?
2: New BN-Embedded Phenanthridines Accessed by the (4 + 2)-BN-Arex Reaction
Given the importance of the v⃗(B–N) vector sense in the context of BN-isosterism,? we also sought an analogue of 4 with an inverted BN unit. The functionalization of the “south side” of the BN-naphthalene 1, i.e. positions α to N, is less developed due to the inability of this site to engage in EAS reaction. Still, we hoped to exploit the selective deprotonation of 1 with strong bases such as t-BuLi ?,? to prepare the new iodinated precursor 2-inv. Since direct lithiation/iodination proved challenging, the target was obtained instead via the trimethylsilyl intermediate 1-SiMe_3_, readily accessible by quenching 1-(4-Li) with Me_3_SiCl (79%), followed by Si-to-I ipso substitution with I_2_ in CH_2_Cl_2_ (Table, bottom panel).
The resulting 2-inv underwent smooth BN-Arex transformation to the target 4-inv in 81% yield? under the standard conditions (Table, bottom panel). The crystal structure of 4-inv confirms the inverted B–N vector with a B–N distance of ≈1.46 Å. The 4-inv remains susceptible to EAS reactivity through its α-boron C-H positions, as confirmed by the synthesis of the iodinated derivative iodo- 4-inv, a compound potentially suitable for further synthetic elaborations.
Following the successful BN-Arex process using aldimines, we proceeded to test the N-silyl ketimines. Gratifyingly, a reaction between the peri-Me substrate 5 and ketimines 17-Me and 17-Bu led to the corresponding C7-alkyl BN-phenanthridines 18 and 19 in 87% and 71% yield, respectively (SchemeA); a somewhat lower yield was obtained for the C7-Ph derivative 20. Curiously, the structural outcome showed a marked dependence on the substituent at the peri position of the iodinated precursor. Indeed, switching from 5 to the nonmethylated precursor 2 in a reaction with 17-Me produced a roughly equimolar mixture of the target BN-phenanthridine 21 and a new species with a molecular mass (m/z = 338), a value consistent with a product bearing an additional norbornene unit (SchemeB). Analogous outcome was also observed for reactions between 2 and the other two ketimines (see Supporting Information, Section 6.1, page S21). Despite having an R f value virtually identical with that of 21, the new species could be isolated in a 32% yield, allowing for its structural determination by NMR.
(A) (4 + 2)-BN-Arex Reaction with N-Silyl Ketimines; (B) Expansion Reaction between Ketimine 17-Me and Substrate 2; (C) Spectroscopic Investigation to Elucidate the Structure of the 8-Membered Ring Adduct 22-b; Key HMBC and NOESY Correlations Are Highlighted in the Planar Structure of 22-b, whilst NOESY Correlations Are Also Highlighted in the 3D Structure; (D) Mechanistic Rationale for the Formation of 22-b; (E) NBDE-mediated (2 + 2 + 4)-BN-Arex Reaction with Alkyl Aryl N-Silyl Ketimines 17-Me and 17-Bu; Formation of New 8-Membered BN Pentacyclic Structures 23 and 24; (F) Access to BN-Dibenzoazepine 25 and BN-Pleiadiene 26
Preliminary analysis led to two possible isomeric 8-membered azocine structures 22-a and 22-b differing in the orientation of their ketamine-derived C–C–C–N fragment. Definitive structural assignment as 22-b was supported by key NOESY and HMBC correlations (SchemeC). HMBC correlations involving the norbornene and the benzoazocine moieties (black arrows), were pivotal, as their relative positioning distinguishes 22-b from 22-a. Long-range HMBC correlations from the norbornane proton H-15 to C-14, C-13, and C-20 were consistent with direct connectivity between the norbornane and benzoazocine units in 22-b. Additionally, a correlation from H-16 to a quaternary carbon resonating at 158.8 ppm, assigned as C-6, was observed. This carbon also showed HMBC correlations from protons from the BN-naphthalene, further supporting structure 22-b exclusively. Furthermore, the NOESY correlations (red arrows) between the aromatic benzoazocine proton H-20 and the norbornane bridgehead methylene protons H-25 and methine H-21 were also fully consistent with 22-b and effectively rule out 22-a. The NOESY data also confirmed the expected cis stereochemistry at the newly formed norbornane junction, in line with 3D model depicted in SchemeC. A unique predominant conformer was found from the DFT geometry optimization as shown in the 3D structure in SchemeC.? Interestingly, compound 22-b is a formal reduced Diels–Alder adduct of an 8-membered azocine ring? that adopts a pseudo boat-like geometry, thus inducing a clear curve shape to the molecule.
The selective conversion of compound 5 to phenanthridines 18–20, as opposed to the divergent conversion of 2 to the phenanthridine/azocine mixture (21 and 22-b), can be rationalized within a unified mechanistic framework that bifurcates depending on the nature of the peri substituent (methyl vs hydrogen). A common Catellani-type Pd/NBE sequence, beginning with oxidative addition of the Nap_BN_–I bond to Pd(0), would thus be followed by norbornene insertion and subsequent oxidative addition of the Ar–Br precursor. This sequence leads to a shared Pd(IV) intermediate,? cross-I, in which an N···Pd interaction with the imine group is postulated?especially considering that desilylation of the imine could already have occurred by this point. As shown in SchemeD, this octahedral Pd(IV) intermediate functions as a mechanistic crossroads, from which the reaction can diverge into two distinct C–C bond-forming pathways: (a) a C(sp^2^)–C(sp^2^) reductive elimination that connects the C2 position of the BN-naphthalene core with the ortho-ketimine carbon, thus forming intermediate phen-II, or (b) a C(sp^2^)–C(sp^3^) bond formation leading to the norbornene-containing intermediate azo-III. Both of these intermediates are expected to undergo intramolecular Buchwald–Hartwig amination, ultimately yielding either BN-phenanthridine 21 or BN-azocine 22-b, respectively. In this framework, the peri-methyl substituent in compound 5 exerts an enhanced ortho effect,? biasing the system toward phen-II and, ultimately, the phenanthridine formation. The diminished steric demand in 2 relaxes this tendency, allowing for a portion of the cross-I population to evolve via azo-III to azocine 22-b. We note that prior mechanistic research highlights the ability of the chelation phenomenon to override the ortho effect, thus enabling the appearance of norbornane-containing derivatives, for example via azo-III. ?,? In case of substrate 5, however, the peri-Me group appears to create a super ortho-effect that brings the reactivity back to the canonical Ar–Ar coupling via phen-II, thus providing the expected phenanthridine targets.
Incidentally, the same reaction conditions also allow for the use of norbornadiene in lieu of norbornene. This enables the coupling between 2 and ketimines 17-Me or 17-Bu to afford BN-azocines 23 and 24 in moderate yields (SchemeE). Despite the potential for these ring systems to undergo a retro-Diels–Alder reaction,? no cyclopentadiene extrusion was observed upon heating 23 and 24 to 130 °C for 16 h. Nevertheless, a related Catellani-style sequence involving norbornadiene incorporation and subsequent cyclopentadiene extrusion, with norbornadiene thus acting as a masked acetylene, was successfully implemented within a (2 + 2 + 3)-BN-Arex process to access new 7-membered derivatives of the BN-naphthalene core. The task was inspired by the scarcity of heptagon rings fused with BN-embedded cores,? which stands in stark contrast with the great potential of such ring system in the design of organic electronics, including through the induction of negative curvatures.? Inspired by an interesting report by Catellani, Derat and co-workers on the synthesis of dibenzoazepine core,? subjecting a mixture of 2 and 2-bromoaniline under catalytic conditions led to the corresponding NBE intermediate, as observed by an ^1^H NMR sampling; further heating to 130 °C enabled smooth evolution of this species to BN-dibenzoazepine 25 via a retro-DA reaction (SchemeF, top). In a related experiment inspired by the work of Kwong et al.,? a decarboxylative coupling between 2 and 8-bromo-1-naphthoic acid initially led to a NBE-containing intermediate that readily evolved to the new BN-extended pleiadiene 26 (61%) upon heating at 130 °C (SchemeF, bottom).
Preparation of New BN-Fluorenone Cores
Seeking to further expand the chemical space of BN-embedded poly heteroarenes via Pd/NBE-enabled ortho/ipso ring fusion, and motivated by the broad utility of fluorenone frameworks? in biological,? photonic, and organic electronic applications,? our next goal was to access fluorenone-type cores through the reaction of 2 or 5 with 2-bromo(hetero)aryl aldehydes.? We were pleased to find that heating a mixture of 2 and 2-bromobenzaldehyde under our standard conditions at 90 °C for 17 h afforded the BN-benzo[a]fluorenone 27 in 43% yield (SchemeA),? along with side products that include 1-norbornyl-BN-naphthalene (see compound S1 in Supporting Information). Interestingly, the earlier improvement in efficiency noted during the formation of the phenanthridine core upon switching from 2 to the methylated iodo BN-naphthalene 5 was also mirrored in this case, with 5 converted to 1-methyl BN-fluorenone 28 in 71% yield. The reaction worked well with bromo-benzaldehyde coupling partners bearing the electron-withdrawing 3-trifluoromethyl group (comp. 29, 50% yield) and the 3-aldehyde group (comp. 30, 66% yield). The process was also extended to the piperonal-derived BN-embedded fluoren-11-one 31 obtained in a 68% yield. The solid-state structure of this compound (see ORTEP drawing in Scheme, center right) revealed a B–N bond length of 1.48 Å, in line with a partially delocalized BN fragment;? the packing diagram also revealed extensive π-stacking of the polyaromatic cores (Figure S13). Given the promise of heteroarene-containing π-cores in the development of organic optoelectronic materials? and new bioactive cores,? the Pd/NBE ArEx reaction was also applied to prepare the BN-embedded tetracyclic benzo[a]thieno-fluorenone 32, as well as nicotinic and picolinic-derived aza-benzo-[a]-fluororenones 33–36 in moderate to good yields (Scheme, A).
(A) (3 + 2)-BN-Arex Reaction with 2-bromo Aryl (or Heteroaryl) Aldehydes: [a] Reaction Conditions: Iodo-BN Substrate (0.2 mmol), 2-bromo Aryl (or Heteroaryl) Aldehyde (0.3 mmol), Pd(OAc)2 (0.010 mmol), Tri(2-furyl)phosphine (0.022 mmol), NBE (1 mmol), and Cs2CO3 (0.6 mmol) in DME (4 mL), 90 °C, 17 h. [b] 0.03 mmol of Pd(OAc)2 Were Utilized; (B) Illustrative Synthetic Elaboration from BN-Fluorenone 27
The newly formed BN-embedded tetracycles derived from 2 preserve a C–H site α to the boron center, rendering them amenable to further functionalization via EAS. This feature is showcased for the BN-fluorenone 27, readily transformed into iodinated derivative 37 in 50% yield (unoptimized) upon exposure to I_2_/AgOTs in CH_2_Cl_2_ (SchemeB). This further enables Pd-catalyzed cross-coupling, with a Sonogashira reaction followed by deprotection furnishing the alkynyl derivatives 38 and 39 in 78% and 67% yields, respectively. The gold(III)-catalyzed hydration of the latter provided the diketone BN-fluorenone 40 in a 60% yield.
How Consequential is the BN-Mapping on Photophysical Properties?
Having secured access to BN isosteres of benzo[c]phenanthridine and benzo[a]fluorenone, we were now well positioned to probe the photophysical consequences of CC-for-BN replacement, including the influence of BN vector orientation. For the benzo[c]phenanthridine framework, the absorption spectrum of the parent (non-BN) carbo-4 (for preparation, see Supporting Information, Section 5) is dominated by two high-intensity bands peaking at 254 and 262 nm, with additional lower-intensity bands with maxima at 342 and 359 nm (FigureA-top, solid black). The fluorescence spectrum mirrors the lowest-energy bands with a negligible Stokes shift, suggesting that the molecule is very rigid both in the ground and in the first excited singlet state. From the crossing point of the overlapping absorption and fluorescence bands, the energy of the first excited singlet state is calculated as 334 kJ mol^–1^. Interestingly, striking spectral differences are immediately apparent for the boron-up BN isostere 4. The highest-energy absorption bands now appear at longer wavelengths and with smaller absorption coefficients, by a remarkable factor of ∼ 2 for the red-most band, while the lowest-energy band shifts to the blue (FigureA, middle, solid red). At the same time, the fluorescence spectrum shifts to the red, resulting in a large Stokes shift (dashed red line). Furthermore, a striking contrast exists between the clearly structured emission observed for carbo-4 and the structureless fluorescence spectrum of 4, suggesting a loss of rigidity in the excited state. The calculated energy of the singlet excited state is now 346 kJ mol^–1^, a value higher than for the parent structure carbo-4. Finally, flipping the orientation of the BN-vector (the boron-down form 4-inv) results in intermediate behavior. A comparison with the all-carbon parent structure carbo-4 showed that inverse-sense BN doping has modest to negligible impact on the lowest-energy absorption bands, the Stokes shift, and the excited singlet state energy. In contrast, the highest-energy absorption region is more similar to that of the boron-up BN isomer 4, and the fluorescence spectrum is also broadened and noticeably less structured. Consistent with the above trends, the fluorescence lifetime (τ_F_) is 5.0 ns for carbo-4, a value that decreases to 3.1 ns for 4, and shows an intermediate lifetime of 4.2 ns for 4-inv (see Table S1). The complete photophysical characterization of the remaining BN-phenanthridines 6, 8–10, 12, 13, 15, 16 and 18–21, as well as new BN-doped 7- and 8-membered cyclic cores 22-b and 23–26 are shown in the Supporting Information (Figures S2–S5).
(A) Absorption (solid lines) and fluorescence emission (dashed lines) spectra of carbonaceous phenanthridine (carbo-4; black), boron-up BN-phenanthridine (4; dark red), and boron-down BN-phenanthridine (4-inv; blue) derivatives in acetonitrile. (B) Absorption (solid lines) and fluorescence (dashed lines) spectra of fluorenone carbo-27 (black), BN fluorenone 27 (red), and 27-inv fluorenone (blue) derivatives in acetonitrile. (C) BN-Arex reaction to prepared 27-inv. Two principal BN-containing byproducts are also depicted. (D) Time-resolved fluorescence kinetic traces of carbo-27 (black), 27 (red) and 27-inv (blue). (E) Fluorescence quantum yield (ΦF) of carbo-27, 27 and 27-inv. (F) 1O2 formation quantum yield (ΦΔ) induced by carbo-27, 27 and 27-inv. As a visual aid, the ΦΔ values for carbo-27, 27, and 27-inv are plotted on logarithmic scale due to significant differences in their values.
Analogously, the benzo[a]fluorenone compound set was also characterized from the optical and photophysical point of view. Hence, the boron-up BN-fluorenone 27 exhibited absorption and fluorescence spectra quite similar to those of the parent CC compound carbo-27, albeit with a slight blue shift apparent in both spectra of 27 (FigureB, top and middle, solid lines for absorption and dashed for emission). In turn, the singlet excited state lifetime of the BN isostere is significantly shorter (τ_F_ = 1.6 ns for 27 vs 3.5 ns for carbo-27; see FigureD), while the fluorescence quantum yield Φ_F_ is comparable (0.006 vs 0.004; FigureE).
Undoubtedly, the ultimate goal of electronic tuning, including through BN mapping, is to obtain molecules with enhanced target function. Cognizant of the general ability of aryl ketones to undergo rapid and efficient singlet-to-triplet intersystem crossing (ISC), we decided to compare the ability of benzofluorenones 27 and carbo-27 to induce the generation of singlet oxygen (^1^O_2_), which occurs from the triplet excited state. Beyond its chemical and biological roles, singlet oxygen production reflects efficient population of the triplet excited state. Indeed, the specific energy ordering of nπ* and ππ* excited states in aromatic ketones strongly affects the fate of the excited states.?
The simplest fluorenone molecule has been shown to exhibit efficient intersystem crossing (ISC) to the triplet state in nonpolar solvents, while this process is highly inefficient in polar solvents.? In fact, literature data and our own measurements indicate that the ISC in carbo-27 is limited in both nonpolar and polar solvents, with internal conversion dominating, particularly in polar solvents.? Not surprisingly, we have found that the quantum yield of singlet oxygen production (Φ_Δ_), an energy-transfer process that occurs from long-lived excited triplet states, is only 0.05 (FigureF, black bar), confirming the triplet state formation being a minor pathway. Remarkably, the Φ_Δ_ value of BN-fluorenone 27 is ∼10-fold higher (0.77; FigureF, red bar), indicating that the singlet-to-triplet ISC pathway is strongly favored by the BN substitution. Assuming that the triplet quantum yield is equal to Φ_Δ_, the intersystem crossing rate constant (k ISC) is calculated as 4.7 × 10^8^ s^–1^ for 27, 23-fold higher than for carbo-27 (2.0 × 10^7^ s^–1^, see Table S3). Of note, the discrepancy between the enhancement of k ISC and Φ_Δ_ indicates that internal conversion must be slower for 27, consistent with the higher energy of its singlet excited state (FigureB-top and middle), on account of the energy-gap law.? As far as we are aware, this is the first case in which enhanced conversion of ^3^O_2_ into ^1^O_2_ is achieved via direct BN-mapping of an all-carbon sensitizer candidate.?
Intrigued by the effect of inverting the orientation of the BN unit on the ^1^O_2_ generation capacity, we sought the “B-down” isomer 27-inv, a species not prepared during the initial synthetic campaign. Although the (3 + 2)-BN-Arex coupling of the BN-inverted iodo precursor 2-inv proved less efficient, the reaction did afford quantities of the target fluorenone 27-inv (FigureC) sufficient for subsequent photophysical characterization. This lower efficiency was partially explained by the formation of an atropoisomeric mixture of BN structure 41 (34%) and the cyclobutyl-fused 42 (36%), the latter likely arising from a direct C–C reductive elimination in the BN-naphthalene-norbornyl-palladacycle BN-NaphNP-A-inv (FigureC, right). In contrast, species 41 might well arise as a consequence of a second metalation event, followed by a reductive elimination, after the successful incorporation of the benzaldehyde electrophile (late intermediate BN-NaphNP**-A-peri**).
The absorption spectrum of 27-inv exhibits essentially the same overall profile as that of the boron-up isomer 27, except that the band system centered at ∼370 nm disappears, leaving only the underlying broader structureless absorption, a band clearly observed in the spectrum of 27. Strikingly, compound 27-inv showed no detectable fluorescence and an almost negligible singlet oxygen Φ_Δ_ value of 0.01 (FigureE, blue bar), even lower than the carbonaceous carbo-27. Thus, the change in the orientation of the boron–nitrogen unit in 27-inv has a dramatic effect on the photophysical properties of the fluorenone core, stripping it of any photochemical activity. The photophysical characterization of the rest of substituted BN-fluorenones (28–40) is shown in the Supporting Information (Figures S6–S9). Φ_Δ_ values range from 0.07 to 0.80, reflecting the nature and position of the substituents in a way consistent with the effects on ^1^O_2_ generation described for substituted fluorenones.?
In this context, interesting insights can be gained from DFT modeling of benzofluorenones carbo-27, 27 and 27-inv at the CAM-b3lyp/Def2TZVP level. As we expected, all three compounds display similarly shaped frontier orbitals, with the HOMO (bottom) and LUMO (top) electron densities illustrated in FigureA. Despite these similarities, the BN doping alters orbital energies, with the −1.56 eV LUMO energy of carbo-27 raised to −1.47 eV in the boron-up BN isostere 27, and lowered to −1.69 eV in the boron-down derivative 27-inv. Given that the LUMO orbital is responsible for accepting an electron upon reduction, this energy perturbation was found to directly translate into the compounds’ reduction potentials.? Hence, the cyclic voltammetry measurements in CH_3_CN for all three species revealed two pseudoreversible reduction waves, presumably signaling the formation of the radical anion [Benzofluorenone] ^ •– ^ and a second doubly reduced species. For the reference carbo-27, the first reduction wave was found at E 1/2 = −1.14 V vs the Standard Calomel Electrode (SCE). In line with the computed LUMO energy ordering (FigureB) this wave shifts the lower potential of E 1/2 = −1.22 V for 27, but in contrast moves up to the higher potential of E 1/2 = −1.03 V for 27-inv (FigureC). Illustrating once again electronic tuning afforded by BN-mapping, these measurements reveal how the redox potential of an organic core can be adjusted - up and down- in a ∼190 mV span by playing with the presence and sense of the BN vector (for full data, see Supporting Information).
*(A) Frontier orbitals shapes and energies (eV) of the benzofluorenone set obtained by DFT calculations at CAM-b3lyp/Def2TZVP level. (B) LUMO energy ordering for carbo-27, 27 and 27-inv. (C) Cyclic voltammograms (reduction region) measured in CH3CN with TBAPF6 electrolyte at 100 mV/s scan rate and referenced to SCE. Partially irreversible wave.
Ultimately, orbital perturbations both in the ground and excited states appear to culminate in 27 significantly outperforming its carbonaceous and boron-down isostere as a triplet sensitizer in ^1^O_2_ production, as was shown in FigureF. Despite some preliminary exploration by TD-DFT,? the exact origin of this phenomenon remains under investigation.
Conclusion
In conclusion, this study shows that earlier advances in BN-naphthalene synthesis pave the way for the construction of larger BN-doped polyarenes via Pd/norbornene-promoted core extension. A Catellani-type aryl extension (ArEx) process is thus configured to engage iodo-BN-naphthalene substrates, affording a plethora of new BN-isosteric architectures, including BN-doped benzo[c]phenanthridines, curved 7- and 8-membered ring-fused derivatives, and BN-embedded benzofluorenones. The newly accessed structural diversity underscores the profound effect of the presence and the directionality of the BN unit on the photophysical properties of the polyarene framework. Most notably, light-induced singlet oxygen (^1^O_2_) generation promoted by the benzofluorenone core shows a 10-fold enhancement in the “boron-up” BN isostere, while dropping to negligible levels upon inversion of the BN unit. These findings could contribute to the development of a new generation of singlet oxygen photosensitizers with altered properties derived from the BN-isosteric replacement.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1h Abengozar, A. ; García-García, P. ; Fernández-Rodríguez, M. A. ; Sucunza, D. ; Vaquero, J. J. Chapter FourRecent developments in the chemistry of BN-aromatic hydrocarbons. In Advances in Heterocyclic Chemistry; Academic Press, 2021; Vol. 135, pp 197–259.
- 2a Knack D. H.Marshall J. L.Harlow G. P.Dudzik A.Szaleniec M.Liu S.-Y.Heider J.BN/CC isosteric compounds as enzyme inhibitors: N- and B-ethyl-1,2-azaborine inhibit ethylbenzene hydroxylation as nonconvertible substrate analogues Angew. Chem., Int. Ed.2013522599260110.1002/anie.201208351 PMC 374881223355270 · doi ↗ · pubmed ↗
- 3a Wang X.-Y.Lin H.-R.Lei T.Yang D.-C.Zhuang F.-D.Wang J.-Y.Yuan S.-C.Pei J.Azaborine compounds for organic field-effect transistors: efficient synthesis, remarkable stability, and BN dipole interactions Angew. Chem., Int. Ed.2013523117312010.1002/anie.20120970623400958 · doi ↗ · pubmed ↗
- 4a Xu S.Zhang Y.Li B.Liu S.-Y.Site-selective and stereoselective trans-hydroboration of 1,3-enynes catalyzed by 1,4-azaborine-based phosphine–Pd complex J. Am. Chem. Soc.201613844145661456910.1021/jacs.6b 0975927802037 PMC 5591746 · doi ↗ · pubmed ↗
- 5a Morgan M. M.Piers W. E.Efficient synthetic methods for the installation of boron–nitrogen bonds in conjugated organic molecules Dalton Trans.2016455920592410.1039/C 5DT 03991 F 26583306 · doi ↗ · pubmed ↗
- 6Rulli F.Sanz-Liarte G.Roca P.Martínez N.Medina V.Puig de la Bellacasa R.Shafir A.Cuenca A. B.From propenolysis to enyne metathesis: tools for expedited assembly of 4a,8a-azaboranaphthalene and extended polycycles with embedded BN Chem. Sci.2024155674568010.1039/D 3SC 06676 B 38638215 PMC 11023045 · doi ↗ · pubmed ↗
- 7a Sun F.Lv L.Huang M.Zhou Z.Fang X.Palladium-catalyzed cross-coupling reactions of 4a,8a-azaboranaphthalene Org. Lett.2014165024502710.1021/ol 502339 h 25226093 · doi ↗ · pubmed ↗
- 8For an example of a non-aromatic cyclizative extension of 1 see:Zhang Y.Sun F.Dan W.Fang X.Friedel–crafts acylation reactions of BN-substituted arenes J. Org. Chem.201782128771288710.1021/acs.joc.7b 0234329083179 · doi ↗ · pubmed ↗