Encephalocraniocutaneous lipomatosis: a rare and sporadic phakomatosis
Ana Clara Maia Palhano, Julia Maria de Oliveira Neumayer, Milene Tiburcio Narenti Ferradoza, Luciana Paula Samorano, Maria Cecilia Rivitti-Machado, Zilda Najjar Prado de Oliveira

Abstract
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
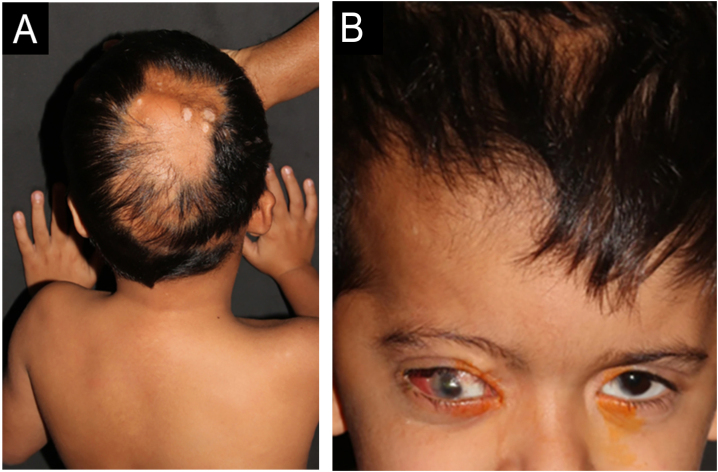 Figure 1
Figure 1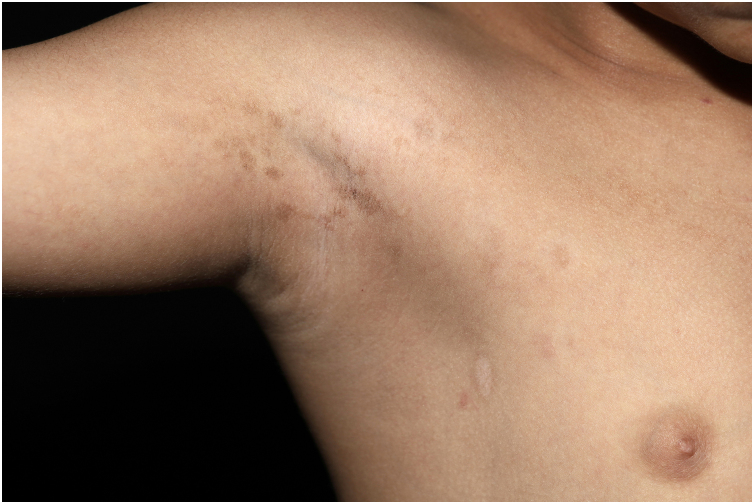 Figure 2
Figure 2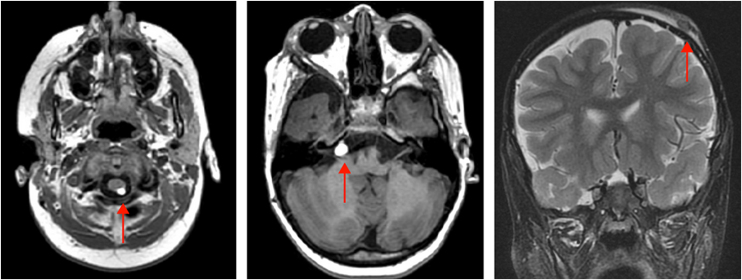 Figure 3
Figure 3Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenetic and rare skin diseases. · Cancer and Skin Lesions · Hedgehog Signaling Pathway Studies
Dear Editor,
Encephalocraniocutaneous Lipomatosis (ECCL), also known as Haberland Syndrome, is a rare and sporadic genetic condition characterized by congenital alterations that simultaneously affect the skin, eyes, and central nervous system.1, 2 This association of manifestations in different systems reflects its inclusion in the group of phakomatoses, a heterogeneous set of genetic diseases that share cutaneous, ophthalmological, and neurological anomalies, frequently accompanied by developmental dysplasias and a predisposition to the emergence of tumors.3
The pathogenesis is attributed to post-zygotic somatic mutations in genes such as FGFR1, KRAS, and NRAS, which promote aberrant activation of the RAS-MAPK signaling pathway. This process results in mosaicism and ectodermal dysgenesis, explaining the asymmetrical distribution and heterogeneity of clinical manifestations.2 Genetic alterations also compromise vasculogenesis, favoring the presence of dysmorphic intracranial vessels, in addition to predisposing to the development of mesenchymal tumors, such as lipomas. One of the most characteristic skin lesions is nevus psiloliparus—a patch of softened alopecia, preferentially located on the scalp, consisting of ectopic mature adipose tissue in the dermis and absence of hair follicles and cutaneous appendages.2, 4
Due to the sharing of the RAS-MAPK pathway with other RASopathies, such as Neurofibromatosis type 1 (NF1), it is possible to observe some similar cutaneous findings in ECCL, such as café-au-lait spots.1, 2 This report identified axillary freckles, a finding not yet documented in cases of ECCL, but which can be explained by the same pathogenic mechanism. The diagnosis of ECCL is challenging due to the wide spectrum of clinical presentations and the rarity of the condition, often requiring a high degree of clinical suspicion and a multidisciplinary approach for confirmation.
A five-year-old male patient was referred to the pediatric dermatology clinic with patches of alopecia on the scalp, especially in the vertex and right parietal regions, accompanied by ocular anomalies present since birth. There was also a history of delayed neuropsychomotor development, episodes of seizures, behavioral changes, and a diagnosis of hydrocephalus at three months of age, requiring a ventriculoperitoneal shunt. In addition, the patient was under ophthalmological follow-up, having already undergone ocular surgery for partial excision of epibulbar lipodermoid—a benign congenital ocular choristoma, that is, a malformation consisting of normal tissue in an anomalous location, in this case, ectopic adipose tissue in the bulbar conjunctiva (Fig. 1).Fig. 1(A) Aplasia cutis over a normochromic alopecia patch in the vertex region; (B) Ipsilateral ocular lipodermoids and low-set right ear.Fig. 1
On physical examination, a heterogeneous alopecic plaque was identified on the scalp, located in the vertex region, with areas of atrophy and others with a softened consistency, similar to adipose tissue, with a clinical appearance compatible with nevus psiloliparus (Fig. 1). The histopathology of one portion of the lesion was suggestive of aplasia cutis. The presence of freckles in the axillary region ipsilateral to the ocular and neurological findings was also observed (Fig. 2). Other anomalies, such as craniosynostosis of the sagittal suture, short stature, low hairline in a "V" shape in the posterior cervical region, low-set right ear with a folded helix, thin lips with eversion of the upper lip, and overlapping fingers, compatible with clinodactyly, were also evidenced. Magnetic resonance imaging (MRI) revealed multiple intracranial lipomas, as well as thickening of the tissue corresponding clinically to the area of alopecia (Fig. 3) in the right frontoparietal region. Based on the clinical picture, histopathological findings, and MRI results, a diagnosis of ECCL was made.Fig. 2. Axillary freckles ipsilateral to ocular and neurological findings.Fig. 2. Fig. 3MRI demonstrating a lipoma in the right pontocerebellar cistern and a lipoma posterior to the cervical spinal cord. Subcutaneous tissue thickening is also observed in the right frontoparietal region, clinically corresponding to the alopecia area.Fig. 3
ECCL represents a distinct clinical and genetic neurocutaneous phenotype, characterized by dermatological, ocular, and neurological alterations. Definitive diagnosis of the disease is based on major and minor criteria, distributed among the three affected systems (Table 1).5, 6 The condition belongs to the group of RASopathies, a set of genetic syndromes caused by mutations in genes that regulate the RAS-MAPK signaling pathway.2 These conditions share similar molecular mechanisms and may show overlapping clinical manifestations. Café-au-lait spots, already described in cases of ECCL, are also present in other RASopathies, such as NF1 and cardiofaciocutaneous syndrome.7 Additionally, axillary freckles were observed, a manifestation not yet documented in association with ECCL, but which could be explained by the same pathogenic mechanism. This pattern is observed in other RASopathies, such as Neurofibromatosis-Noonan syndrome, NF1, and Legius syndrome, reinforcing the possibility of phenotypic overlap mediated by activation of the RAS-MAPK pathway.7, 8Table 1ECCL.Table 1. SystemMajor CriteriaMinor CriteriaPatient FindingsEyes
-
1Choristoma, with or without associated anomalies.
-
2Corneal or other anterior chamber anomalies
-
3Ocular or palpebral coloboma
-
4Calcification of the eyeball Choristoma (major), coloboma (minor)Skin
-
1Confirmed nevus psiloliparus (NP)
-
2Possible NP and ≥ 1 of the minor cutaneous criteria (criteria 2–5)
-
3≥2 minor criteria among criteria 2–5
-
1Irregular or banded alopecia, non-scarring
-
2Subcutaneous lipoma(s) in the frontotemporal region
-
3Focal aplasia/hypoplasia of the skin on the scalp
-
4Small nodular lesions on the skin of the eyelids or between the outer corner of the eye and the tragus Possible NP (major), subcutaneous lipomas in the frontotemporal region, non-scarring alopecia, focal skin aplasia (confirmed by biopsy)Central Nervous System (CNS)
-
1Intracranial lipoma
-
2Intraspinal lipoma
-
3≥2 minor criteria
-
1Abnormal intracranial vessels
-
2Arachnoid cyst or other meningeal abnormality
-
3Complete or partial atrophy of a cerebral hemisphere
-
4Porencephalic cyst
-
5Asymmetrically dilated ventricles or hydrocephalus
-
6Calcifications (except in the basal ganglia) Intracranial lipoma (major), volumetric reduction of the right frontal and temporal lobes, diffuse prominence of cerebrospinal fluid spaces on the right side, small arachnoid cyst in the right middle cranial fossa.Others
-
1Jaw tumor (osteoma, odontoma, or ossifying fibroma)
-
2Multiple bone cysts
-
3Coarctation of the aorta N/AN/ADefinitive Case.1. Involvement of 3 systems, with major criteria in ≥ 2 of them; or.2. Involvement of 3 systems, with confirmed or possible nevus psiloliparus (NP) and ≥ 1 of the minor cutaneous criteria (criteria 2–5); or.3. Involvement of 2 systems with major criteria, one of which must include confirmed or possible nevus psiloliparus and ≥ 1 of the minor cutaneous criteria (criteria 2–5).
Differential diagnoses include Oculocerebrocutaneous Syndrome (Delleman Syndrome), Proteus, Epidermal Nevus and Schimmelpenning Syndrome.7, 9, 10 The therapeutic strategy depends on the symptoms. Endoscopic ventriculostomy of the third ventricle is generally preferred for relief of intracranial hypertension in cases of hydrocephalus.11 Surgery is recommended for symptomatic spinal lipomas, aiming to preserve neurological function, with follow-up being essential to monitor the progression of lipomatous lesions and prevent vertebral deformities.11
ORCID IDs
Julia Maria de Oliveira Neumayer: 0009-0001-9651-1942
Milene Tiburcio Narenti Ferradoza: 0000-0002-5864-7259
Luciana Paula Samorano: 0000-0001-7077-8553
Maria Cecilia Rivitti-Machado: 0000-0003-2910-7330
Zilda Najjar Prado de Oliveira: 0000-0002-8596-1999
Authors' contributions
Ana Clara Maia Palhano: Design and planning of the study; collection, analysis, and interpretation of data; drafting and editing of the manuscript; approval of the final version of the manuscript.
Julia Maria de Oliveira Neumayer: Analysis and interpretation of data; drafting and editing of the manuscript; approval of the final version of the manuscript.
Milene Tiburcio Narenti Ferradoza: Analysis and interpretation of data; drafting and editing of the manuscript; approval of the final version of the manuscript.
Luciana Paula Samorano: Clinical intervention in the case; critical review of the manuscript; approval of the final version of the manuscript.
Maria Cecilia Rivitti-Machado: Clinical intervention in the case; critical review of the manuscript; approval of the final version of the manuscript.
Zilda Najjar Prado de Oliveira: Research orientation; critical review of the manuscript; approval of the final version of the manuscript.
Ethical approval and informed consent
The study was conducted in accordance with the institution's ethical standards, and informed consent was obtained from the patient's legal guardian.
Approval statements
All authors read and approved the final version of the manuscript.
Consent for publication
Written informed consent was obtained from the patient's legal guardian for the publication of this case report and corresponding images.
Financial support
None declared.
Research data availability
Not applicable.
Conflicts of interest
None declared.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Subbiah D.Asiff N.Hamzah N.Samsudin A.Encephalocraniocutaneous lipomatosis: a case report and literature review Cureus 142022 e 3249810.7759/cureus.32498 PMC 983760136644099 · doi ↗ · pubmed ↗
- 2Richters R.Seyger M.Meeuwis K.Rinne T.Eijkelenboom A.Willemsen M.Oculoectodermal syndrome – encephalocraniocutaneous lipomatosis associated with NRAS mutation Acta Derm Venereol 1002020 adv 0012110.2340/00015555-3358 PMC 923494031633190 · doi ↗ · pubmed ↗
- 3Ruggieri M.Polizzi A.Marceca G.P.Catanzaro S.PraticòA.D.Di Rocco C.Introduction to phacomatoses (neurocutaneous disorders) in childhood Childs Nerv Syst 362020222922683294077310.1007/s 00381-020-04758-5 · doi ↗ · pubmed ↗
- 4Happle R.Küster W.Nevus psiloliparus: a distinct fatty tissue nevus Dermatology 1971998610969317810.1159/000017968 · doi ↗ · pubmed ↗
- 5Sofiatti A.A.Cirto A.G.Arnone M.Romiti R.Santi C.Leite C.Encephalocraniocutaneous lipomatosis: clinical spectrum of systemic involvement Pediatr Dermatol 23200627301644540710.1111/j.1525-1470.2006.00165.x · doi ↗ · pubmed ↗
- 6Siddiqui S.Naaz S.Ahmad M.Khan Z.A.Wahab S.Rashid B.A.Encephalocraniocutaneous lipomatosis: a case report with review of literature Neuroradiol J 3020175785822870796110.1177/1971400917693638 PMC 5703133 · doi ↗ · pubmed ↗
- 7Jafry M.Sidbury R.RA Sopathies Clin Dermatol 3820204554613297260310.1016/j.clindermatol.2020.03.010 · doi ↗ · pubmed ↗
- 8Lopes F.C.Schroeder C.Patel B.Levy M.L.Review of encephalocraniocutaneous lipomatosis Semin Pediatr Neurol 52202410116610.1016/j.spen.2024.10116639622606 · doi ↗ · pubmed ↗