Identification and functional characterization of a novel nonsense mutation of CASR gene in a familial hypocalciuric hypercalcemia pedigree
Yang Tian, Bingyang Liu, Yang Xudan, Ruojun Qiu, Zhang Shaojun, Huang Haoshu, Wu Fang, Fenping Zheng

Abstract
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
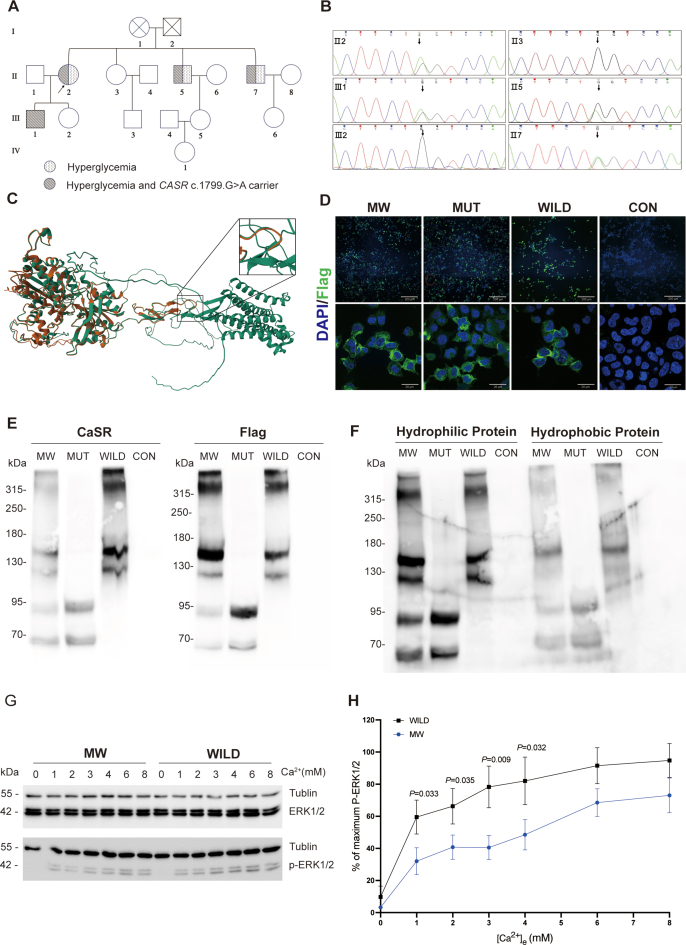 Figure 1
Figure 1Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsParathyroid Disorders and Treatments · Bone health and treatments · Medical Imaging and Pathology Studies
Familial hypocalciuric hypercalcemia (FHH), a rare cause of hypercalcemia, features a benign, lifelong mild-to-moderate hypercalcemia. It is often misdiagnosed as primary hyperparathyroidism, so patients may wrongly undergo parathyroidectomy. Thus, it is important to raise the suspicion of FHH before the diagnosis of primary hyperparathyroidism. Familial hypocalciuric hypercalcemia 1 (FHH1) is caused by heterozygous inactivating mutations of calcium-sensing receptor (CaSR) gene, and CaSR is a 1078-amino acid G-protein-coupled receptor and is mostly expressed in the parathyroid gland and renal tubule to maintain the homeostasis of calcium by regulating parathyroid hormone secretion and urinary calcium excretion. So far, over 150 different germline mutations of CASR in FHH1 have been reported, with 85% being missense mutations and only 3%–4% being nonsense mutations.1^,^2 In this study, we report a novel heterozygous nonsense mutation on the CASR (c.1799 G > A, p.Trp600∗) gene; this variant is predicted to cause an amino acid alteration from tryptophan to a premature stop codon in the extra-cellular domain, and the affected proband presented with mild hypercalcemia, low calcium to creatinine clearance ratio, and hyperglycemia with reserved insulin secreting function.
We firstly conducted pedigree analysis including clinical manifestations, laboratory tests characteristics (Fig. 1A; Table S1), and mutation site of CASR gene verification (Fig. 1B), and our results confirmed this nonsense mutation in the CASR gene segregated with the clinical traits of FHH1; we also found two siblings of the proband, who harbored CASR mutation, were also afflicted with hyperglycemia with reserved insulin secreting function (Table S2).Figure 1A novel nonsense mutation of CASR gene is associated with FHH1. (A) Pedigree of a family with FHH1. The squares represent male and the circles represent female family members. Diabetes mellitus-affected individuals are represented by speckled symbols, and hypercalcemia together with CASR mutant are represented by slash shading symbols. The index case is marked by an arrow. (B) Sanger sequencing results of exon 7 of the CASR gene in the family members. Subjects Ⅱ2, Ⅱ5, Ⅱ7, and Ⅲ1 carried the heterozygous CASR mutation and concurrently exhibited hypercalcemia, which demonstrates that the phenotype segregates with the genotype within this pedigree. (C) Effect of CASR mutation on the modeled structure of the calcium sensing receptor. The red and green ribbons represent mutant and wild monomeric CaSR, respectively, compared with the wild-type c.1799 G > A, which caused a truncated protein in the extracellular domain of the CaSR receptor. (D) Cellular expression of Flag in transfected HEK293T cells. Confocal microscopy images showed that Flag (green) was localized to the periphery of MW, MUT, and WILD, but not in CON. Nuclei were labeled with DAPI (blue). Scale bar: 10 μm. The Flag signals were confirmed at the membrane periphery. (E) Western blotting analysis of total protein. In the MW and MUT groups, two distinct bands were detected below 70 KDa and 95 KDa, corresponding to Flag and CaSR proteins, respectively. In contrast, these bands were not observed in the WILD and CON groups. (F) Western blotting analysis of hydrophilic and hydrophobic proteins. In the western blotting profiles of hydrophobic proteins from the MW and MUT groups, two relatively faint bands were identified below 70 KDa and 95 KDa. This observation indicates the expression of truncated protein forms on the cell membrane surface. (G) MAPK activity response to eCa^2+^. MW showed a blunted response to eCa^2+^ compared with WILD. (H) Effect of different eCa^2+^ concentrations on phosphorylation of ERK1/2 of MW and WILD. Concentrations of eCa^2+^ (0, 1, 2, 3, 4, 6, 8 mM) were plotted on the x-axis, the ratio of pERK1/2 over total ERK1/2, expressed as a percentage of the maximum response value for each concentration measured based on band intensity, was on the y-axis. Subsequently, the two curves were compared (values were plotted as mean ± standard error of the mean; p-values indicate the unpaired MW t-test on WILD for each concentration). The data presented were the cumulative results of four independent biological replicates. CASR, calcium sensing receptor; FHH1, familial hypocalciuric hypercalcemia 1; MW, HEK293 cells transfected with 1 μg mutant CASR plasmid together with 1 μg wild CASR plasmid; MUT, HEK293 cells transfected with 2 μg mutant CASR plasmid; WILD, HEK293 cells transfected with 2 μg wild CASR plasmid; CON, HEK293 cells transfected with 2 μg of control plasmid; MAPK, mitogen-activated protein kinase; ERK1/2, extracellular signal-regulated kinase 1/2.Figure 1
According to the American College of Medical Genetics and Genomics (ACMG) guideline, this nonsense mutation was predicted to be pathogenic. An online SWISS-MODEL workspace (https://swissmodel.expasy.org) was employed to predict the effect of this variant on the protein structure of CaSR, demonstrating that this alteration will lead to a truncated CaSR protein in the extracellular domain (Fig. 1C), with a predicted molecular weight of 66.04 kDa.
Because a large proportion of nonsense mutations are prone to undergo nonsense-mediated decay, we performed an in vitro study of this mutation to assess membrane expression. We constructed a 3flag-tagged human CASR W600∗ plasmid through overlap PCR and transfected HEK293 cells with 1 μg mutant CASR plasmid together with 1 μg wild CASR plasmid (MW) to mimic heterozygous mutation of CASR, 2 μg mutant CASR plasmid (MUT), 2 μg wild CASR plasmid (WILD), and 2 μg of control plasmid (CON). Western blotting of total protein extracted from the four groups using anti-CaSR (ThermoFisher: MA1-934, corresponding to amino acids 215–235 of human CaSR) and anti-Flag antibody showed immunoreactive bands lower than 70 kDa and 95 kDa in MW and MUT corresponding to the truncated monomeric form of CaSR (Fig. 1E). Hydrophobic and hydrophilic proteins extracted and enriched by CelLytic™ MEM Protein Extraction Kit (Sigma–Aldrich: CE0050) were also subjected to western blotting, and faint immunoreactive bands lower than 70 kDa and 95 kDa in hydrophobic protein indicate the membrane expression of this truncated CaSR (Fig. 1F). Fluorescence immunocytochemistry assay in MW, MUT, and WILD displayed similarly specific staining using anti-Flag antibody (green), while cells in CON showed no green fluorescence signal, confirming cell membrane expression of flag-tagged CaSR (Fig. 1D). We then accessed mitogen-activated protein kinase (MAPK) responsiveness to investigate transduction activity of CaSR,3 in both MW and WILD group, and observed evident phosphorylation of extracellular signal-regulated kinase 1/2 (ERK1/2) when the eCa^2+^ was elevated from 0 to 8 mM but a blunted phosphorylation response of ERK1/2 of the MW group compared with the WILD group (Fig. 1G and H). This indicated that the heterozygous mutation of CASR W600∗ resulted in hypoactivation of the receptor and therefore failed to maintain the homeostasis of calcium.
We also reviewed CASR mutations in FHH1. So far, 14 distinct nonsense mutations, including our case, have been reported. Among them, 8 out of 14 mutations are located in exon 7, with an average S–Ca of 2.827 ± 0.047 mM (presented as mean ± standard error of the mean). The other 6 mutations occur in exons 2–4, are predicted to undergo nonsense-mediated decay, and show milder hypercalcemia (average S–Ca 2.625 ± 0.024 mM) (Table S3). The CaSR exists as homodimers on the cell surface. Its extracellular domain forms a Venus flytrap structure and contains 5 calcium-binding sites (CaBS-1 to 5), and its dimerization is essential for normal CaSR function. In nonsense mutations, premature stop codons located 50–55 bp upstream of the final exon–exon junction will be subjected to nonsense-mediated decay, causing haploinsufficiency and a milder phenotype. In contrast, premature stop codons in the gene's 3′ region, including the last exon and ∼55 bp of the penultimate, evade nonsense-mediated decay, yielding truncated proteins. These truncated proteins disrupt normal CaSR dimerization, exerting a dominant-negative effect. This mechanism explains the variable severity of hypercalcemia caused by CASR nonsense mutations at different genomic locations. This might partly explain the lower incidence of nonsense mutations in FHH1 compared with autosomal dominant hyperparathyroidism, as the mild phenotype caused by a nonsense mutation subjected to nonsense-mediated decay could be overlooked.
Although FHH1 has long been regarded as a benign and non-threatening condition, an in-depth data analysis by Ridge Dershem4 reveals that CASR variants are associated with a range of health issues including cardiovascular problems such as embolism, thrombosis, and chronic diastolic heart failure, as well as neurological disorders including dementia, major depression, pancreatitis, and a propensity for alcohol abuse, underlining the need for a more comprehensive understanding and vigilant monitoring of FHH1 and its related gene variants. Interestingly, within this family, our findings revealed that, except for the younger patient III:1, the CASR W600∗ harboring individuals also exhibited type 2 diabetes mellitus with reserved insulin secretion. This is notable because relatively few prior studies have documented the association between FHH1 and hyperglycemia. Insulin is the major hypoglycemic hormone. Previous investigations on small cohorts of individuals with FHH1 have documented that their glucose tolerance test outcomes and insulin release test profiles exhibit no significant difference from the general population, and glucagon-like peptide-1 (GLP-1), secreted by intestinal L-cells, also plays a crucial role in regulating glucose homeostasis. Within the family pedigree reported by our research team, three diabetic patients can be categorized as the GLP-1 secretion-deficient subtype according to the refined classification of type 2 diabetes mellitus proposed by Zou Dajin. Both calcium ions and amino acids, particularly aromatic amino acids, function as strong agonists of the CaSR. CaSRs localized on the intestinal epithelium are capable of modulating GLP-1 secretion by sensing calcium ions and amino acids within the intestinal lumen. The marked reduction up to 70% in GLP-1 secretion induced by CaSR antagonists, such as Calhex 231 and NPS2143, and the substantial increase in GLP-1 secretion mediated by the CaSR agonist calindol, collectively underscore the indispensable role of intestinal CaSR in maintaining blood glucose homeostasis.5 The alteration of the spatial structure of the CaSR weakens the secretion of GLP-1 mediated by calcium ions and amino acids, which may partly account for the hyperglycemia observed in the three individuals with CASR mutations within this pedigree. However, since we have not carried out further investigations into insulin, GLP-1, and glucose-dependent insulinotropic polypeptide secretion, we are unable to draw a definite conclusion regarding the relationship between this nonsense mutation and hyperglycemia.
In conclusion, we reported a novel nonsense mutation, c.1799 G > A, in the CASR gene within an FHH1 pedigree. Subsequent in vitro functional analyses revealed that this germline mutation led to a truncated form of CaSR and blunted the reaction of MAPK pathway. However, it remains ambiguous whether this genetic variant plays a role in the development of hyperglycemia.
CRediT authorship contribution statement
Yang Tian: Writing – review & editing, Writing – original draft, Project administration, Methodology, Investigation, Funding acquisition, Formal analysis, Data curation, Conceptualization. Bingyang Liu: Writing – original draft, Methodology, Data curation, Conceptualization. Yang Xudan: Methodology. Ruojun Qiu: Data curation. Zhang Shaojun: Writing – original draft, Conceptualization. Huang Haoshu: Methodology, Conceptualization. Wu Fang: Writing – review & editing, Methodology, Investigation, Data curation, Conceptualization. Fenping Zheng: Writing – review & editing, Writing – original draft, Data curation, Conceptualization.
Ethics declaration
The study was approved by the Ethics Committee of the Fourth Affiliated Hospital, Zhejiang University School of Medicine (K2021125). All participants provided written informed consent after a detailed explanation of the study protocol.
Funding
This work was supported by the 10.13039/501100014759Medical Scientific Research Foundation of Zhejiang Province, China (No. 2022KY197).
Conflict of interests
The authors declared no conflicting interests.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Hannan F.M.Kallay E.Chang W.Brandi M.L.Thakker R.V.The calcium-sensing receptor in physiology and in calcitropic and noncalcitropic diseases Nat Rev Endocrinol 151201833513044304310.1038/s 41574-018-0115-0PMC 6535143 · doi ↗ · pubmed ↗
- 2Hannan F.M.Nesbit M.A.Zhang C.Identification of 70 calcium-sensing receptor mutations in hyper- and hypo-calcaemic patients: evidence for clustering of extracellular domain mutations at calcium-binding sites Hum Mol Genet 21122012276827782242276710.1093/hmg/dds 105 · doi ↗ · pubmed ↗
- 3Zhang C.Miller C.L.Brown E.M.Yang J.J.The calcium sensing receptor: from calcium sensing to signaling Sci China Life Sci 581201514272557645110.1007/s 11427-014-4779-y · doi ↗ · pubmed ↗
- 4Dershem R.Gorvin C.M.Metpally R.P.R.Familial hypocalciuric hypercalcemia type 1 and autosomal-dominant hypocalcemia type 1: prevalence in a large healthcare population Am J Hum Genet 106620207347473238655910.1016/j.ajhg.2020.04.006PMC 7273533 · doi ↗ · pubmed ↗
- 5Li R.J.W.Barros D.R.Kuah R.Small intestinal Ca SR-dependent and Ca SR-independent protein sensing regulates feeding and glucose tolerance in rats Nat Metab 61202439493816772610.1038/s 42255-023-00942-4 · doi ↗ · pubmed ↗