Familial colorectal cancer: search for novel predisposition genes
Asta Försti, Beiping Miao, Abhishek Kumar, Dagmara Dymerska Zaremba, Magdalena Marciniak, Jan Lubinski, Kari Hemminki

TL;DR
This study searches for new genes that may cause inherited colorectal cancer by analyzing families with multiple cases.
Contribution
The study identifies novel candidate genes and pathways involved in inherited colorectal cancer predisposition.
Findings
Cell cycle/DNA repair and TGFβ signaling pathways were found to be significantly associated with CRC predisposition.
Variants in APCDD1, CYBA, PTK7, and SRC genes were identified in multiple families.
Most variants were family-specific, suggesting a polygenic inheritance pattern.
Abstract
Family history of colorectal cancer (CRC) and multiple primary CRCs in a single person may indicate inherited CRC predisposition. In the present study, we performed whole exome/genome sequencing on germline DNA from at least two CRC cases in 19 families and from family members with a double primary CRC from seven additional families. We used a set of in silico predictions in combination with a STRING protein–protein interaction and pathway analysis to identify the most likely variants predisposing to CRC. We identified Cell cycle/DNA repair and TGFβ signaling/Focal adhesion/Extracellular matrix organization pathways as highly significant protein–protein interaction networks. Variants in the APCDD1, CYBA, PTK7 and SRC genes were identified in more than one family, and they were shown to dysregulate basic cellular functions, potentially leading to cancer development. Most variants were…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
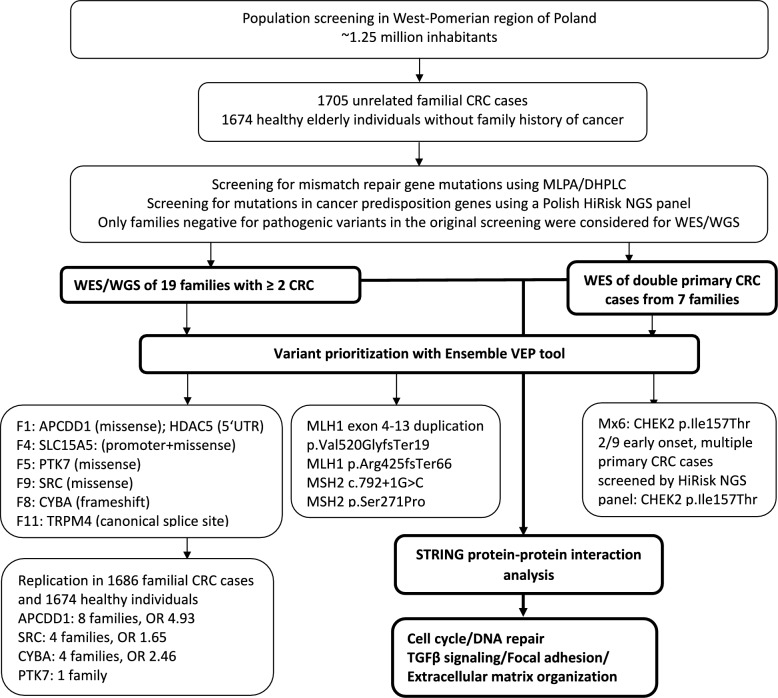 Figure 1
Figure 1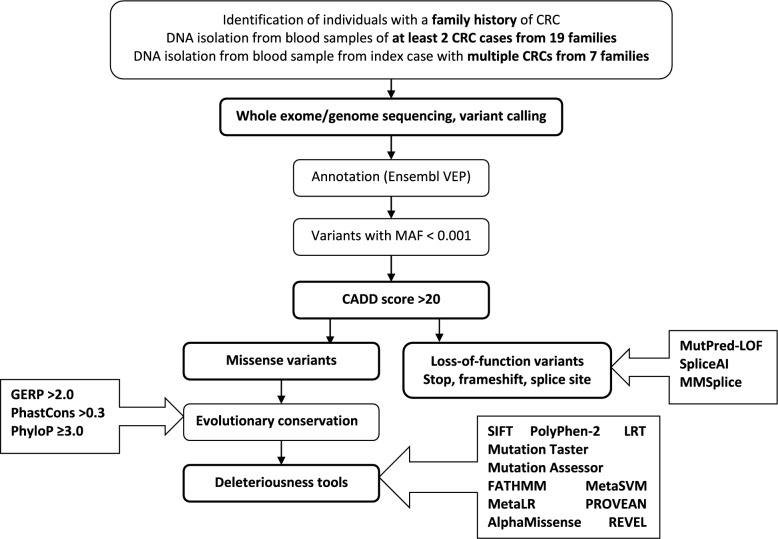 Figure 2
Figure 2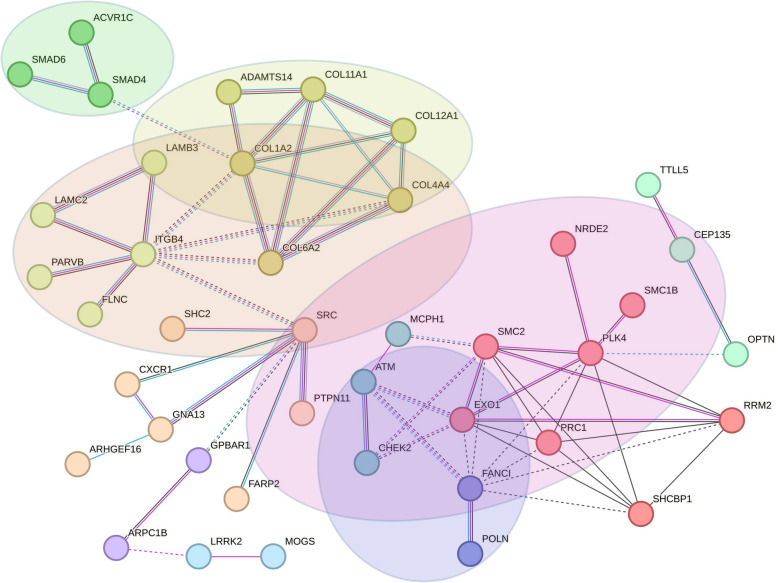 Figure 3
Figure 3- —Deutsches Krebsforschungszentrum (DKFZ) (1052)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenetic factors in colorectal cancer · Colorectal Cancer Treatments and Studies · BRCA gene mutations in cancer
Background
Family history of colorectal cancer (CRC) and multiple primary CRCs in a single person may indicate inherited CRC predisposition. Familial risk of CRC increases from 1.70 when one family member is diagnosed with CRC to 2.76 when two or more family members are diagnosed [1]. Familial CRC also increases the risk of double primary CRCs, up to 5 times compared to the sporadic disease [2]. The well-established high-risk CRC predisposition genes include the mismatch repair (MMR) genes MSH2, MLH1, MSH6 and PMS2 causing Lynch syndrome, and the Wnt-signaling gene APC causing familial adenomatous polyposis syndrome. In addition, variants in the base-excision repair gene MUTYH are causing recessively inherited adenomatous polyposis syndrome. Other well-characterized genes associated to dominantly inherited polyposis syndromes include TGFβ-pathway genes SMAD4 and BMPR1A, in addition to the AMP-activated protein kinase STK11 [3]. During the last 10 years, genome-wide sequencing has promoted studies attempting to identify novel high-to-moderate cancer predisposition genes [3]. These attempts have been successful in identifying genes predisposing to CRC polyposis syndromes, including polymerase proofreading genes POLE and POLD1 [4], base excision repair gene NTHL1 [5], MMR gene MSH3 [6], TGFβ-pathway gene GREM1 [7] and Wnt-pathway gene RNF43 [8]. However, variants in these genes are rare, and they do not explain familial clustering of non-polyposis CRC.
In Sweden, about 15% of CRC patients have a first degree relative diagnosed with CRC [1], but according to a UK sequencing study of familial patients only about one quarter were explained by variants in the known high-penetrance genes [9]. Recently, genome-wide sequencing efforts, including our own, have attempted to fill in this gap and they have provided evidence for novel high-to-moderate penetrance CRC predisposition genes [3, 10–15]. Most of these genes need confirmation in additional families as well as functional validation of the identified variants. Here, we summarize and report our published and unpublished data on potential CRC predisposition genes after using an update of our Familial Cancer Variant Prioritization Pipeline (FCVPP) [16] in combination with a STRING protein–protein interaction and pathway analysis [17] on 19 CRC families, from which at least two cases were available for whole exome/genome sequencing (WES/WGS) and on seven additional CRC families presenting with double primary CRCs. We provide a list of all genes and variants prioritized by our pipeline to help scientists in the CRC predisposition community in further validation.
Materials and methods
Population
In the West-Pomeranian region of Poland, about 1.25 million (~ 70%) inhabitants participated in a population screening that was performed mainly in years 2000–2001. Cancer family history was collected from the participants and persons with positive CRC family history were invited to outpatient clinics of the Hereditary Cancer Center, Department of Genetics and Pathology, Pomeranian Medical University, Szczecin, Poland and their family histories were collected through face-to-face detailed interviews. An average review took 20–30 min. Similarly, persons with negative cancer family history were interviewed. Eligible individuals were asked to participate to the study and they signed an informed consent. Altogether, 1705 unrelated familial CRC cases and 1674 healthy elderly individuals without family history of cancer agreed to participate to the study and provided a blood sample for germline variant analysis. The study participants were screened for MMR gene mutations using multiplex ligation-dependent probe amplification (MLPA) and denaturing high-performance liquid chromatography (DHPLC), and more recently using a Polish HiRisk next generation sequencing (NGS) panel for mutations in the genes APC, ATM, BRCA1, BRCA2, CDH1, CDKN2A, CHEK2, MLH1, MUTYH, MSH2, MSH6, NBN, PALB2, PTEN, PMS2, RAD51C, RAD51D, STK11, TP53.
For the WES/WGS study, only families negative for the known cancer predisposition mutations based on the methodology used at the time of blood sample collection and initial screening were considered. From 19 families, at least two family members who were affected by CRC agreed to participate as described earlier [14]. Additionally, seven independent index cases with double primary CRCs were included in the study as described [15]. All these families had a prominent family history of CRC with the earliest age at diagnosis ranging from 23 to 64 years (Supplementary Fig. 1).
Whole exome/genome sequencing (WES/WGS)
Germline DNA extracted from peripheral blood samples was used for WES/WGS. WES of families F1–F5 was performed using the Agilent SureSelect V5 with UTR target capture kit on the Illumina HiSeq 2000 platform and WGS of families F6–F15 and FA-FD was performed using the Illumina TrueSeq Nano DNA kit on the Illumina HiSeqX10 V2.5 platform at the DKFZ core facility as described [10, 14]. We converted the variant positions from the human reference genome GRCh37/hg19 to GRCh38/hg38 using the Assembly Converter of Ensembl release 114 -May 2025. The seven independent double primary CRCs were whole-exome sequenced by BGI Genomics using the Illumina Agilent V6-based sequencing (https://www.bgi.com/global) and the reads were aligned to the GRCh38/hg38 human reference genome as described [15].
Variant annotation and filtering
Variants were annotated and filtered using an update of our in-house developed FCVPP [16]. We annotated the identified single nucleotide variants (SNVs) and small insertion/deletions (InDels) that were present in all affected family members of each family or in the double primary CRC cases using the Ensembl Variant Effect Prediction (VEP) tool [18]. We included only variants affecting the Ensembl canonical protein coding transcript. Minor allele frequency (MAF) filter of 0.1% was used with respect to gnomAD_exomes_AF, gnomAD_exomes_NFE_AF and gnomAD_exomes_POPMAX_AF data to remove common variants (gnomAD v.4.0.0; https://gnomad.broadinstitute.org). Rare SNVs and InDels ranking within the top 1% of potentially deleterious variants in the human genome were selected using the Combined Annotation Dependent Depletion (CADD v.1.7) tool; a scaled PHRED-like CADD score greater than 20 was applied [19].
Variant prioritization: missense variants
We used several in silico tools to identify the most likely pathogenic missense variants. We screened the variants for their potential deleteriousness by using 11 different prediction tools: Sorting Intolerant from Tolerant (SIFT) [20], Polymorphism Phenotyping version 2 (PolyPhen-2) [21], Log ratio test (LRT) [22], MutationTaster [23], Mutation Assessor [24], Functional Analysis Through Hidden Markov Models (FATHMM) [25], MetaSVM [26], MetaLR [26], Protein Variation Effect Analyzer (PROVEAN) [27], AlphaMissense [28] and REVEL [29]. Variants predicted to be deleterious by > 50% of these tools were selected. To evaluate whether the variants are located in an evolutionary conserved position, we used three tools: Genomic Evolutionary Rate Profiling (GERP > 2.0) [30], PhastCons (> 0.3) [31] and Phylogenetic P-value (PhyloP ≥ 3.0) [32], with an inclusion cutoff of at least two positive predictions. To evaluate whether the affected genes are intolerant to variation, we report the Z-score, developed by the gnomAD consortium for missense variants [33]. Positive Z-scores indicate increased constraint, i.e. intolerance to variation.
Loss-of-function variant analysis
Stop-gain, frameshift and splice-site variants affecting the canonical splice sites were considered when the CADD score criterion of > 20 was met. Variants affecting the last exon of the gene were excluded from further analyses, all other variants are reported. In order to discriminate pathogenic and neutral variants, we used MutPred2 (http://mutpred.mutdb.org) [34]. For each variant, it renders a score between zero and one; higher scores denote variants that are more likely to be pathogenic; a conservative threshold score of 0.50 at 5% false positive rate is recommended. MutPred2 also shows structural and functional mechanisms that are impacted in the affected region of the protein, accompanied by significant prior-corrected P values. Splice site variants were analyzed by using SpliceAI [35] and MMSplice [36] within the CADD v.1.7 tool. The SpliceAI Δ score ≥ 0.5 indicates a confidently predicted cryptic splice variant and Δ score ≥ 0.8 indicates a high-scoring predicted cryptic splice variant. The MMSplice score (absolute value) > 2 indicates a high confidence level prediction and > 1.5 medium level confidence. For LoF variants we report the LOEUF (loss-of-function observed/expected upper bound fraction) score from gnomAD which reflects the gene constraint; gnomAD recommends to use LOEUF < 0.6 as an indication of increased gene constraint, if a cut-off is needed, although many tumor suppressor genes, such as the MMR genes have a higher LOEUF.
Network analysis with STRING
Protein–protein interactions within a biologically relevant pathway may give information about the importance of the identified genes in CRC development. We used the in silico tool STRING [17] to investigate the interactions between the proteins encoded by the prioritized genes and protein–protein interaction (PPI) enrichment within the created network. To create the network we used experiments, databases and co-expression as active interaction sources with a minimum required interaction score 0.400. In STRING, the PPI enrichment analysis is based on the input data compared to the random list of proteins. Such an enrichment indicates that the proteins are at least partially biologically connected, as a group. To calculate the false discovery rate (FRD), STRING uses the Benjamini–Hochberg procedure. We visualized the networks as clusters of interacting proteins in order to analyze functions that are specific to each cluster. For that purpose, we used Markov clustering (MCL) [37] as suggested by STRING and we report corresponding functional description of the clusters and the genes within the clusters. We also investigated pathway enrichment of the proteins within the two largest interacting networks based on Biological Processes within Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG) and Reactome. Further information of genes, proteins and their function were collected using GeneCards (https://www.genecards.org) and UniProt (https://www.uniprot.org). PubMed (https://pubmed.ncbi.nlm.nih.gov) was used to search for information about the relationship between the genes with cancer, especially in the context of CRC.
Results
Families and variant prioritization
The workflow of our study is shown in Fig. 1. We searched for rare, potentially pathogenic variants in 19 independent families, from which at least two CRC cases were sequenced using WES or WGS, and in seven independent cases with a double primary CRC and a family history of CRC using WES (Supplementary Fig. 1). The families were previously screened for pathogenic variants in known CRC predisposition genes using methodologies available at the time of recruitment of the CRC cases. When we screened the WES/WGS data for known CRC predisposition genes [38], we found three families with nonsense mutations in the MMR genes, including a large duplication of exons 4–13 causing an amino acid change p.Val520GlyfsTer19, a frameshift variant (p.Arg425SerfsTer66) and a canonical splice donor variant in intron 4, respectively [39]. We also found a missense MSH2 variant p.Ser271Pro in one additional family. In one double primary CRC case, Mx6, we found a moderate penetrance variant in CHEK2, p.Ile157Thr [15].Fig. 1. Workflow of the study. The integrated analysis of all variants prioritized using the Ensembl VEP analysis and the STRING protein–protein interaction analysis as the main focus of the study are highlighted in bold
In our primary analysis for identification of novel CRC predisposition genes we excluded the families with the MMR gene mutations. We prioritized the variants that segregated with CRC in the 15 remining families or that were present in the double primary CRC cases using the pipeline shown in Fig. 2. We identified altogether 258 variants that passed the criteria of the pipeline, including 177 missense (89 in families/88 in double primary CRC cases), 31 stop-gain (17/14), 13 canonical splice site (3/10) and 37 frameshift (15/22) variants (Supplementary Tables 1 and 2). In the families F1-F15, there were between 1 (F3) and 22 (F12) variants per family; in the double primary CRC cases, the numbers were slightly higher, between 11 (Mx46) and 27 (Mx7).Fig. 2. Pipeline for identification of the germline variants potentially predisposing to CRC
STRING protein–protein interaction and pathway analysis
To further prioritize the most likely candidates for CRC predisposition genes, we combined the gene lists from the families and the double primary CRC cases, and used the corresponding protein list as an input for the STRING protein–protein interaction (PPI) analysis. Altogether 91 proteins interacted with at least one other protein (PPI enrichment P value 6.33 × 10^−7^) (Supplementary Fig. 2). MCL clustering analysis identified 32 clusters (Supplementary Table 3). From these, six clusters were connected to each other in a large interaction network related to KEGG pathways “TGFβ signaling”, “Focal adhesion” and GO Biological process “Extracellular matrix organization” (Fig. 3). More specifically, the MCL algorithm-based clusters were related to “Signaling by Activin” (ACVR1C, SMAD4, SMAD6), “Collagen biosynthesis and modifying enzymes” (COL1A2, COL4A4, COL6A2, ADAMTS14, COL12A1, COL11A1), “Cell junction organization” (ITGB4, PARVB, FLNC, LAMC2, LAMB3), “Netrin mediated repulsion signals” (SRC, FARP2, SHC2, CXCR1, GNA13, PTPN11, ARHGEF16) and two undetermined two-protein clusters (ARPC1B, GPBAR1 and LRRK2, MOGS) (Table 1). Another larger network was related to GO Biological process “Cell cycle” and Reactome pathway “DNA repair” (Fig. 3) and included the MCL algorithm-based clusters “SMC proteins Flexible Hinge Domain” (PLK4, RRM2, SHCBP1, NRDE2, SMC1B, SMC2, EXO1, PRC1), “DNA damage induced protein phosphorylation” (ATM, MCPH1, CHEK2), “Fanconi Anemia Pathway” (FANCI, POLN) and one unclassified cluster of three proteins (CEP135, OPTN, TTLL5) (Table 1). Both the proteins identified in the families and in the double primary CRC cases contributed to these large networks, as shown in Supplementary Fig. 2.Fig. 3STRING protein–protein interaction networks of proteins encoded by the prioritized genes in 15 families and seven double primary CRC cases. Only the two largest networks are shown. Purple line between the nodes indicates experimental evidence of interaction between the proteins, light blue line indicates database evidence and black line co-expression evidence. Proteins related to KEGG pathway “TGFβ signaling” are shown in a green circle, “Focal adhesion” in an orange circle and GO Biological process “Extracellular matrix organization” in a yellow circle. Proteins related to GO Biological process “Cell cycle” are shown in a pink circle and Reactome pathway “DNA repair” in a blue circleTable 1Description of genes and variants in the main STRING clustersFamily_IDGeneGene nameChrom_Pos_Ref_AltEnsemble transcript; HGVScHGVSpCADD_phredDeleteriousness score n/11MutPred2_LOF scoreMutPred2 mechanismsProtein functionFamily 1FLNCFilamin C7_128840655_G_AENST00000325888.13:c.1657G > AENSP00000327145.8:p.Gly553Ser25.59Actin cytoskeleton organization; sarcomere organizationPLK4Polo Like Kinase 44_127896849_GT_GENST00000270861.10:c.2754delENSP00000270861.5:p.Val919CysfsTer42330.43NACentriole replication during cell cycleFamily 5GNA13G Protein Subunit Alpha 1317_65053567_T_CENST00000439174.7:c.445A > GENSP00000400717.2:p.Ile149Val20.87Transmembrane signalingFamily 6LAMB3Laminin Subunit Beta 31_209630647_G_AENST00000356082.9:c.911C > TENSP00000348384.3:p.Pro304Leu27.67.5Cell adhesion; cell migrationCXCR1C-X-C Motif Chemokine Receptor 12_218164374_G_AENST00000295683.3:c.838C > TENSP00000295683.2:p.Arg280Cys25.46.5IL8 signaling; immune responseFamily 7ATMATM Serine/Threonine Kinase11_108301732_G_TENST00000675843.1:c.5262G > TENSP00000501606.1:p.Lys1754Asn236DNA damage response (DSB repair)Family 8COL6A2Collagen Type VI Alpha 2 Chain21_46114063_G_AENST00000300527.9:c.791G > AENSP00000300527.4:p.Arg264His328Cell adhesionFamily 9SRCSRC Proto-Oncogene, Non-Receptor Tyrosine Kinase20_37394253_G_AENST00000373578.7:c.529G > AENSP00000362680.2:p.Val177Met319Cell adhesion; cell cycle; immune responseFamily 12COL11A1Collagen Type XI Alpha 1 Chain1_102995999_C_TENST00000370096.9:c.2285G > AENSP00000359114.3:p.Arg762Gln28.99Extracellular matrix proteinEXO1Exonuclease 11_241889544_G_TENST00000366548.8:c.2485G > TENSP00000355506.3:p.Glu829Ter430.50NADNA mismatch repair; immune responseFamily 14CEP135Centrosomal Protein 1354_56019469_A_CENST00000257287.5:c.3129A > CENSP00000257287.3:p.Lys1043Asn23.46Centriole biogenesisPARVBParvin Beta22_44131621_G_AENST00000338758.12:c.511G > AENSP00000342492.6:p.Val171Met26.97.5Cell adhesion; cell migrationPOLNDNA Polymerase Nu4_2229168_G_AENST00000511885.6:c.64C > TENSP00000435506.1:p.Gln22Ter37NANADNA repair; homologous recombinationFamily 15ARHGEF16Rho Guanine Nucleotide Exchange Factor 161_3477968_CGGCCAACGTGCTACCTTTTCCTGTT_CENST00000378378.9:c.1568_1592delENSP00000367629.4:p.Arg523ProfsTer7350.31NAUnknown; protein–protein and protein–lipid interactionsMx6LAMC2Laminin Subunit Gamma 21_183227656_C_TENST00000264144.5:c.1427C > TENSP00000264144.4:p.Thr476Met23.76.5Cell adhesionCHEK2Checkpoint Kinase 222_28725099_T_CENST00000404276.6:c.470 T > CENSP00000385747.1:p.Ile157Thr23.56.5Cell cycle, DNA repairSMAD4SMAD Family Member 418_51058218_CTGGT_CENST00000342988.8:c.762_765delENSP00000341551.3:p.Gly255SerfsTer8029.50.69NATumor suppressor; TGFbeta signalingSHC2SHC Adaptor Protein 219_460954_CG_CENST00000264554.11:c.42delENSP00000264554.4:p.Ala15ArgfsTer164230.41Methylation (p = 0.037); Amidation (p = 0.044)Signal transductionMx7COL12A1Collagen Type XII Alpha 1 Chain6_75102661_G_TENST00000322507.13:c.8351C > AENSP00000325146.8:p.Pro2784His278Cell adhesion; ECM organizationSMC1BStructural Maintenance Of Chromosomes 1B22_45372273_C_TENST00000357450.9:c.2078G > AENSP00000350036.4:p.Arg693His24.67Cell cycle; meiosisRECQLRecQ like helicase12_21486541_G_AENST00000444129.7:c.439C > TENSP00000416739.2:p.Gln147Ter380.61Magnesium binding (p = 0.036); Amidation (p = 0.040)DNA repairSHCBP1SHC binding and spindle associated 116_46604439_G_AENST00000303383.8:c.712C > TENSP00000306473.3:p.Arg238Ter360.4NA–Cell proliferation, growth and differentiationMx8COL4A4Collagen Type IV Alpha 4 Chain2_227007353_C_TENST00000396625.5:c.5045G > AENSP00000379866.3:p.Arg1682Gln27.18ECM organizationNRDE2NRDE-2, Necessary For RNA Interference, Domain Containing14_90292787_C_GENST00000354366.8:c.1752G > CENSP00000346335.3:p.Glu584Asp23.86.5Cell cycle; DNA damage response; mRNA processingPRC1Protein Regulator Of Cytokinesis 115_90979162_A_CENST00000394249.8:c.1103 T > GENSP00000377793.3:p.Phe368Cys327.5Cell cycleSMAD6SMAD Family Member 615_66703558_TGGGAGCTCCCTGCTGGACGTGGCGGAGCC_TENST00000288840.10:c.306_334delENSP00000288840.5:p.Ser102ArgfsTer928.30.53NABMP and TGF-beta/activin-signalingMx29RRM2Ribonucleotide Reductase Regulatory Subunit M22_10128903_C_TENST00000304567.10:c.854C > TENSP00000302955.4:p.Ser285Leu22.97Cell cycle; deoxyribonucleotide synthesisMx34ACVR1CActivin A Receptor Type 1C2_157556276_G_CENST00000243349.13:c.361C > GENSP00000243349.7:p.Pro121Ala21.56TGFbeta signalingARPC1BActin Related Protein 2/3 Complex Subunit 1B7_99389957_G_TENST00000646101.2:c.445G > TENSP00000496599.1:p.Asp149Tyr327Actin polymerization; transcription regulation; DNA repairSMC2Structural Maintenance Of Chromosomes 29_104118330_A_GENST00000374793.8:c.1951A > GENSP00000363925.3:p.Thr651Ala26.110.5Cell cycle; mitosisADAMTS14ADAM Metallopeptidase With Thrombospondin Type 1 Motif 1410_70752124_C_TENST00000373207.2:c.2626C > TENSP00000362303.1:p.Arg876Cys29.76.5Collagen degradationLRRK2Leucine Rich Repeat Kinase 212_40340400_G_AENST00000298910.12:c.6055G > AENSP00000298910.7:p.Gly2019Ser28.210.5Multiple processes, e.g. autophagy, differentiation; Wnt signalingPTPN11Protein Tyrosine Phosphatase Non-Receptor Type 1112_112477722_A_GENST00000351677.7:c.925A > GENSP00000340944.3:p.Ile309Val236Multiple cell functions, e.g. cell growth, differentiation, mitosis, oncogenic transformationOPTNOptineurin10_13112464_T_TAGENST00000378747.8:c.381_382insAGENSP00000368021.3:p.Asp128ArgfsTer2222.30.30Coiled coil (p = 0.022); Helix (p = 0.045); Carboxylation (p = 0.046)Autophagy; innate immunityFARP2FERM, ARH/RhoGEF And Pleckstrin Domain Protein 22_241468263_CCT_CENST00000264042.8:c.2021_2022delENSP00000264042.3:p.Leu674GlnfsTer3126.60.39Allosteric site (p = 0.037)RAC signal transduction; cell adhesionMx36MOGSMannosyl-Oligosaccharide Glucosidase2_74461994_C_GENST00000448666.7:c.1795G > CENSP00000410992.3:p.Asp599His26.28N-linked oligosaccharide processingCOL1A2Collagen Type I Alpha 2 Chain7_94427840_C_TENST00000297268.11:c.3481C > TENSP00000297268.6:p.Arg1161Cys29.59ECM organizationITGB4Integrin Subunit Beta 417_75756515_C_TENST00000200181.8:c.4795C > TENSP00000200181.3:p.Arg1599Cys316.5Cell adhesion; cancerFANCIFA complementation group I15_89295009_C_TENST00000310775.12:c.2551C > TENSP00000310842.8:p.Gln851Ter430.48NADNA repairMCPH1Microcephalin 18_6442069_T_TCENST00000344683.10:c.586dupENSP00000342924.5:p.Gln196ProfsTer825.50.32Iron binding (p = 7.1352e − 05); Catalytic site (p = 0.0006); Disulfide linkage (p = 0.00156); Proteolytic cleavage (p = 0.005); Signal helix (p = 0.0055)Chromosome condensation and DNA damage induced cellular responsesTTLL5Tubulin tyrosine ligase Like 514_75683658_T_CENST00000298832; HGVSc:c.371 + 2 T > C**33TranscriptionMx43GPBAR1G Protein-Coupled Bile Acid Receptor 12_218262944_CT_CENST00000519574.2:c.221delENSP00000430202.1:p.Leu74ArgfsTer4225.10.30NABile acid receptor^^The tools used to evaluated the deleteriousness of the variants include SIFT, PolyPhen-2, LRT, Mutation Taster, Mutation Assessor, FATHMM, MetaSVM, MetaLR, Provean, Alphamissense and REVEL^**^SpliceAI-don-loss 0.99; MMSp_donor -1.363
Most of the variants in the two main networks were missense variants (Supplementary Tables 1 and 2). However, both SMAD4 (Mx6) and SMAD6 (Mx8) variants affecting the TGFβ signaling pathway caused a frameshift, followed by premature termination of the translation. Within the Cell cycle/DNA repair pathway clusters, three stop-gain variants were identified: EXO1 (F12), FANCI (Mx36) and POLN (F14). Furthermore, two frameshift variants leading to a premature stop codon were found: MCPH1 (MX36) and PLK4 (F1). In eight families, two or more variants within these pathways were prioritized, including F1 with the FLNC and PLK4 variants, F12 with the EXO1 and COL11A1 variants, F14 with the POLN, CEP135 and PARVB variants, Mx6 with the CHEK2, SMAD4, LAMC2 and SHC2 variants, Mx7 with 2 missense variants in SMC1B and COL12A1, Mx8 with the SMAD6, PRC1, NRDE and COL4A4 variants, Mx34 with four missense variants in ACVR1C, ADAMTS 14, PTPN11 and SMC2, and Mx36 with FANCI, MCPH1, ITGB4 and COL1A2 variants.
Other potential predisposition genes
We found also other interesting genes that were related to the pathways identified by the STRING analysis, but not connected to the two main STRING networks. ANKRD53 (stop-gain variant, CADD 39), CDK18 (splice donor-loss variant, CADD 34), KDM8 (CADD 29.2), ZW10 (CADD 26.5), TTC28 (CADD 33) and TUBG2 (CADD 25.6) were among the genes related to cell cycle, and ENDOV (stop-gain variant, CADD 35), POLL (CADD 29.1) and RECQL (stop-gain variant, CADD 38) were related to DNA repair (Supplementary Tables 1 and 2). LTBP3 (CADD 25.3) and TGFBI (CADD 23.5) were related to TGFβ pathway and CDH4 (CADD 25.3), LGALS3BP (stop-gain variant, CADD 34) and PTK7 (CADD 22.7) to cell adhesion (Supplementary Tables 1 and 2).
Previously, we had done a detailed analysis, including basic functional tests, of six individual families and prioritized the Wnt-pathway gene APCDD1 and the histone deacetylase HDAC5 (5’UTR variant) in F1 [11], the innate immunity-related gene SLC15A5 (a missense and a promoter variant) in F4 [13], the Wnt-pathway and cell adhesion gene PTK7 in F5 [10], the proto-oncogene SRC in F9 [12], as well as CYBA (frameshift variant) in F8 and TRPM4 (splice acceptor-loss variant) in F11 related to mucus biology and inflammation [14]. From the encoded proteins, only SRC was present in the main STRING networks (Fig. 3; Table 1). In addition to the SRC variant, which was found in altogether five families, the variant in APCDD1 was found in nine families, the one in CYBA in five families and the one in PTK7 in two families (Supplementary Table 4).
In our secondary analysis we included the genes prioritized by our pipeline in the four families with MMR gene mutation (Supplementary Table 5) to the above STRING analysis. The main contribution of the corresponding proteins to the PPI network was the modification of the Cell cycle/DNA repair cluster, in which MLH1 and MSH2 interacted with each other and with ATM, EXO1 and FANCI (Supplementary Fig. 3, Supplementary Table 6). MSH2 interacted additionally with CHEK2 and SMC2. MSH2 also added RECQL with a stop-gain variant from the double primary CRC case Mx7 to the DNA repair cluster, and connected the Cell cycle/DNA repair cluster to the TGFβ signaling/Focal adhesion/Extracellular matrix (ECM) organization cluster.
Discussion
Only a small proportion of familial CRC is explained by established high-penetrance cancer predisposition genes. World-wide efforts using genome-wide sequencing approaches in CRC families have produced some success in identifying novel predisposition genes for CRC polyposis syndromes, and suggested candidate genes for familial CRC in general [3]. However, most of the candidates have been identified only in single families. In the present study, we combined the data from our WES/WGS studies on 26 CRC families, and identified Cell cycle/DNA repair and TGFβ signaling/Focal adhesion/ECM organization pathways as commonly affected across the families. The variants in the APCDD1, CYBA, and SRC genes were identified in more than one family with odds ratios ranging from 1.6 to 4.9; PTK7 variant was found in another family with four CRC cases [10–12, 14]. Functional studies gave clues for their functional significance.
Among the families we sequenced, we identified three with an MMR gene mutation leading to a truncated protein; in one additional family the MMR gene mutation was a missense amino acid change with unknown significance. These mutations might have been missed due to the methodology, DHPLC, used in the early 2000 [39], highlighting the importance of re-screening historically negative cases with up-to-data technologies. Among the families, from which the double primary CRC case was sequenced, we found a moderate-penetrance variant CHEK2 p.Ile157Thr; this family had also been screened using DHPLC [15]. Interestingly, previous screening using the HiRisk NGS panel had identified two other double primary CRC cases with the same variant, thus suggesting its involvement in CRC predisposition in this type of families [15].
In the Cell cycle/DNA repair pathway, we identified several genes which were affected by variants leading to protein truncation, including EXO1, FANCI, POLN and RECQL with a stop-gain variant and MCPH1 and PLK4 with a frameshift variant. Defects in DNA repair genes are common both in the hereditary and sporadic CRC, highlighted by the well-established role of germline mutations in the MMR genes in Lynch syndrome. Interestingly, our STRING PPI analysis showed both experimental, database and co-expression-based interactions between the MMR genes and genes in our Cell cycle/DNA repair cluster. From our candidate genes, ATM, EXO1 and RECQL were among the genes expressed in the intestine with variants having a CADD score > 20 and MAF < 0.1% in a Swedish study on 55 CRC families [40]. One study has also reported a rare, homozygous missense variant in ATM in an early-onset CRC case, however, acknowledging the problems evaluating the pathogenicity of the large number of rare missense variants identified in ATM [41]. In addition to RECQL, other RecQ like helicases, including WRN and BLM, have been suggested as familial CRC predisposition genes [42, 43]. Also, enrichment of variants in Fanconi anemia DNA repair genes (FANCC, FANCD1, FANCE, FANCJ) in six out of 40 Spanish families has been reported [44] and a biallelic inactivation of FANCM in an advanced CRC case [45]. A study on 48 familial CRC cases from Australia identified a pathogenic stop-gain variant in FANCI, but did not prioritize the gene as it was not linked to cancer in cancer-associated databases COSMIC, OncoKB and TSGene [46]. That study also reported a stop-gain variant with unknown significance in a cancer-associated FANCD2 gene. These studies give further confidence for the involvement of additional DNA repair genes in CRC predisposition.
In the TGFβ signaling/Focal adhesion /ECM organization pathway, frameshift variants were identified in the SMAD4 and SMAD6 genes, as well as in the SHC2 gene. From these, SMAD4 is known to predispose to Juvenile polyposis syndrome [3], but it is also commonly mutated in sporadic CRC [47]. Similar to the SMAD proteins, SHC adaptor proteins are signal transducers mediating signals from growth factors, cytokines and integrins [48]. They have been linked to various diseases, including cancer, however, SHC2 seems to function mainly in the peripheral nervous system [48]. We identified also several missense variants in genes coding both major collagenous and non-collagenous constituents of basement membrane of the ECM, including collagens and laminins, and a missense variant in an integrin-coding gene, ITGB4, involved in the cell-ECM interactions. An abnormal TGFβ-ECM axis has been recognized as an important component in cancer development and progression, promoting cell proliferation, differentiation, survival and migration [49]. Our results highlight a role of germline variation in remodeling the ECM through the TGFβ-focal adhesion-ECM axis.
Some limitations should be considered when interpreting our results. For seven out of the 22 families without MMR mutations, the prioritization of the variants was based only on in silico tools with partly overlapping prediction algorithms. This strategy may have led to identification of false positives. However, for the other 15 families, segregation of the variants with CRC supported their potential pathogenicity, with functional characterization of the prioritized variants from five families. Also, the variants in APCCD1, CYBA, PTK7 and SRC were found in other independent familial CRC cases. However, we acknowledge that although some of the families were large, we had only 2–4 samples from CRC patients from each family available for sequencing, and for the seven double primary CRC families only the index case was available. Additionally, we excluded the few healthy family members who agreed to participate to the study as they usually were from a younger generation, and thus uninformative for the segregation analysis. In the study, we performed both WES and WGS, and used tree different library preparation kits and sequencing platforms, which may have introduced some batch effects. However, as we focused on only coding germline variants, for which each allele should be present at about 50% frequency, the problem should be smaller than in the identification of somatic mutations.
Conclusions
Our study identified the Cell cycle/DNA repair and TGFβ signaling/Focal adhesion/ECM organization as the main pathways affected by germline variants in 26 Polish families. Variants in the APCDD1, CYBA, PTK7 and SRC genes were identified in more than one family, and they were shown to dysregulate basic cellular functions, potentially leading to cancer development. Most variants were private to a family, and each family had more than one candidate variant, thus suggesting a synergistic or polygenic mode of inheritance. This hypothesis, as well as validation of the identified variants, and pathways and their functional consequences, need confirmation by other family-based studies.
Supplementary Information
Supplementary Material 1. Supplementary Material 2. Supplementary Material 3. Supplementary Material 4. Supplementary Material 5. Supplementary Material 6. Supplementary Material 7.