Bromination Functionalization of Diazo Compounds with CBr4 via Convergent Paired Electrolysis
Qian Wang, Wentian Wu, Haibo Mei, Jorge Escorihuela, Romana Pajkert, Gerd-Volker Röschenthaler, Jianlin Han

TL;DR
This paper introduces a new electrochemical method to create α-bromo phosphonates using diazo compounds and CBr4 under mild conditions.
Contribution
The first example of direct anodic oxidation of diazos coupled with in situ-generated nucleophiles via convergent paired electrolysis.
Findings
The method achieves up to 96% yields of α-bromo phosphonates.
It uses mild conditions and shows good substrate compatibility.
The reaction is scalable and introduces a new electrochemical mode for diazo compounds.
Abstract
An electrochemical bromination of diazo compounds with CBr4 as a bromine source via convergent paired electrolysis has been developed, which affords α-bromo phosphonates as products in up to 96% yields. This work represents the first example of direct anodic oxidation of diazos to couple with an in situ-generated nucleophile from the cationic reduction of CBr4 by using CH2Cl2 as a hydrogen source. This reaction features mild conditions, good substrate compatibility, and scale-up applicability, which represents a new electrochemical reaction mode of diazo compounds and also provides easy access to α-bromo phosphonates.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
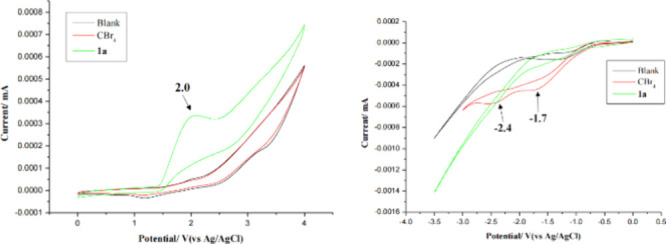 Figure 1
Figure 1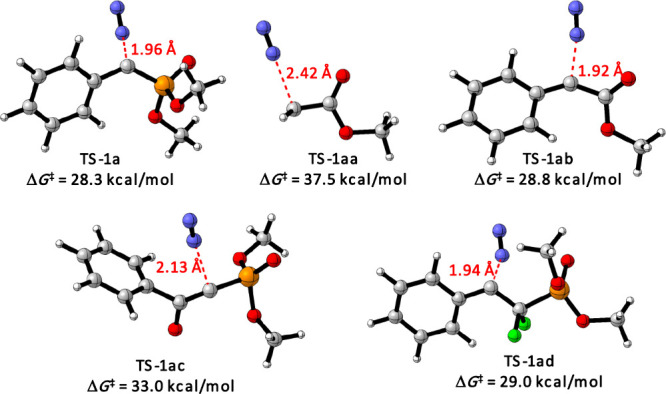 Figure 2
Figure 2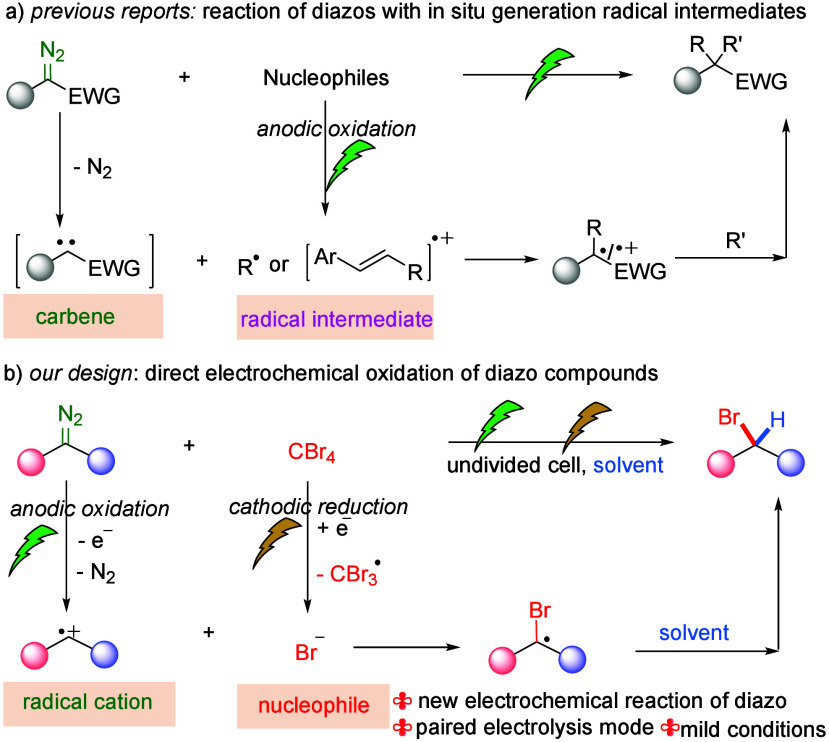 Figure 3
Figure 3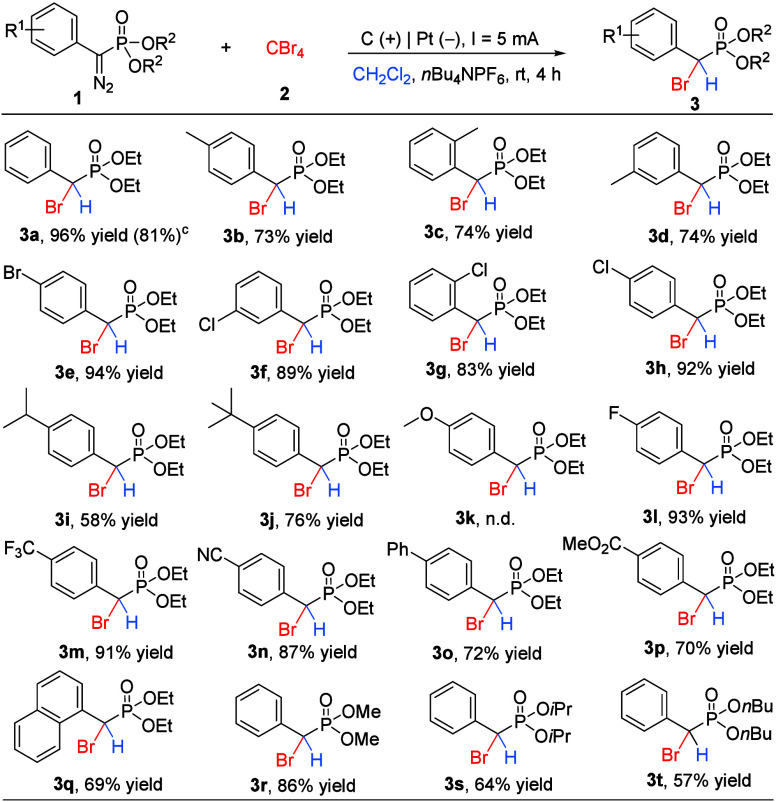 Figure 4
Figure 4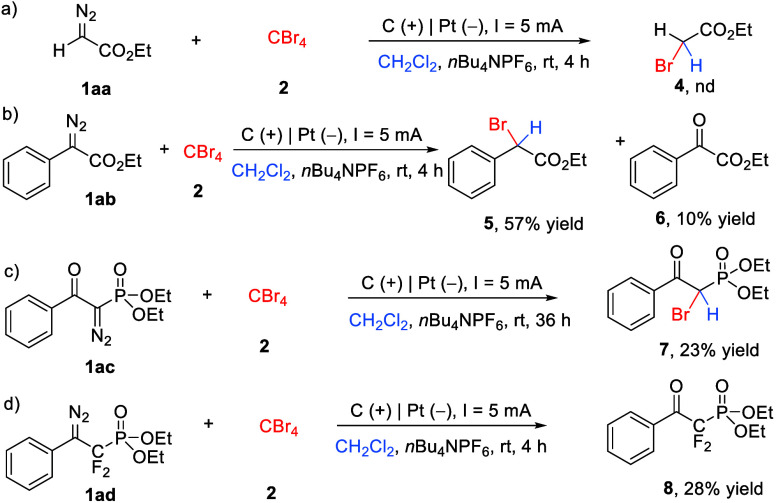 Figure 5
Figure 5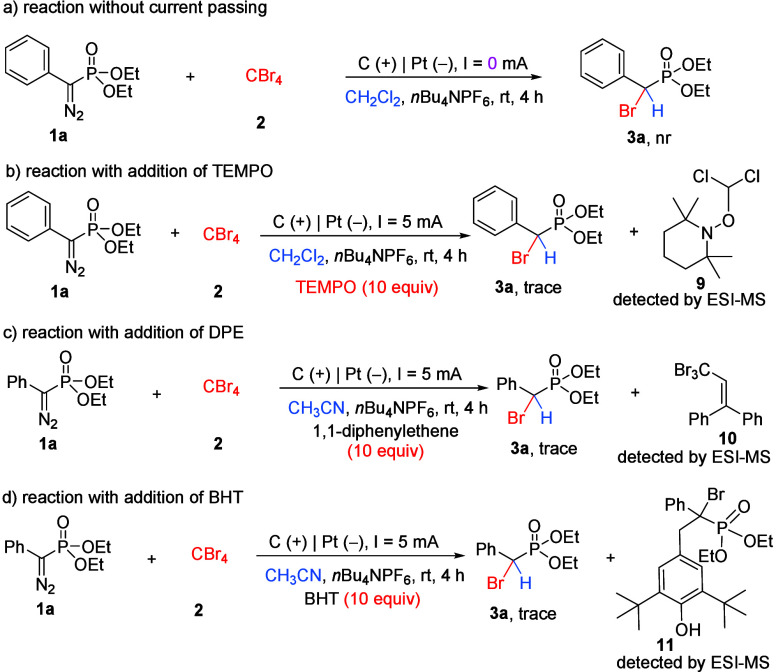 Figure 6
Figure 6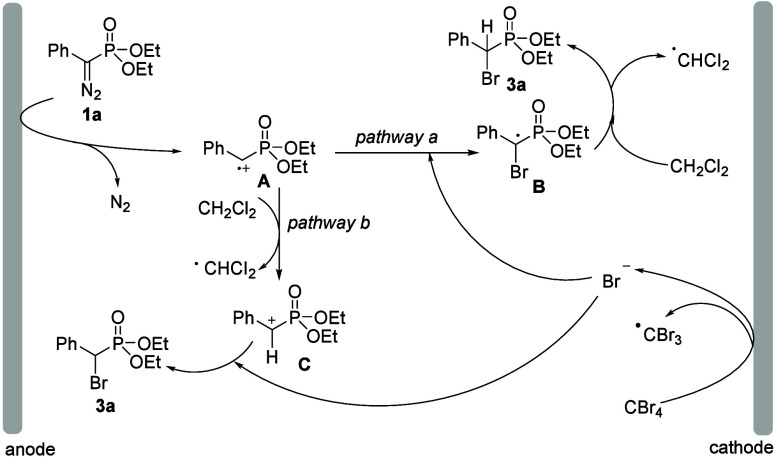 Figure 7
Figure 7 Figure 8
Figure 8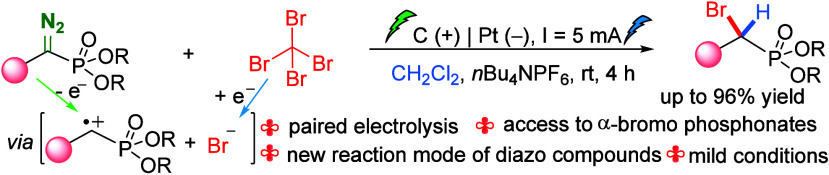 Figure 9
Figure 9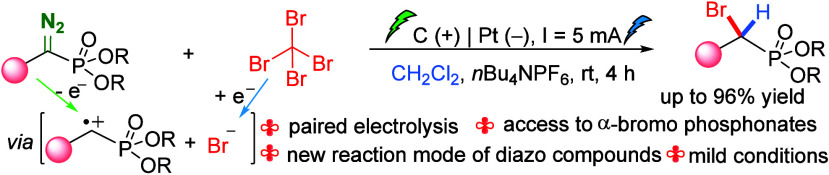 Figure 10
Figure 10Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsVanadium and Halogenation Chemistry · Environmental remediation with nanomaterials · Cyclopropane Reaction Mechanisms
Diazo compounds belong to an extremely important type of organic building blocks, which are found in many natural products? and bioactive molecules.? Diazo compounds feature privileged functional units and can act as powerful and versatile reagents with diverse reactivity patterns.? In recent decades, diazo compounds have found extensive applications in organic synthesis, polymer synthesis, medicinal chemistry, material science, and various other fields. As a result, continuous efforts have been devoted to the development of transformations involving these organic compounds in past decades. ?−? ?
Among various transformations employing diazo compounds, electrochemical multicomponent reactions of diazo compounds have attracted increasing attention in recent years, which is due to the elimination of the need for external oxidants or reductants and the efficiency of generating higher molecular complexity under mild and sustainable conditions.? In 2022, the Huang group developed an electrochemical carbenoid insertion reaction of diazo compounds with thiols and salicylic acids. The reaction proceeded via anodic oxidation of thiols into sulfur radicals, followed by coupling with free carbene from α-diazoester to afford the corresponding products.? In 2023, the Lei group developed an interesting electrochemical difunctionalization of diazo compounds with two different nucleophiles via the anodic oxidation of thiols or N-methyl anilines to generate radical intermediates.? Later in 2024, an electrochemical selenidation-triggered difunctionalization of diazo compounds with varieties of nucleophiles was reported, which proceeded via anodic oxidation of diselenides, affording a diverse array of selenium-containing pyrazole esters and alkoxy esters as products.? The electrochemical cycloaddition reaction between alkenes and diazo esters was also developed, which proceeded via anodic oxidation of olefins followed by a [2 + 1] cycloaddition with diazo compounds toward cyclopropane synthesis.? Although some elegant works have been developed, these reactions focus on diazo compounds acting as radical acceptors (Schemea).? To the best of our knowledge, no electrochemical reaction via direct anodic oxidation of diazo compounds has been explored until now.
In view of the above limitations ?−? ? ? ? and in continuation of our interest in electrochemical transformations? and diazo compound functionalization,? we envision that electrochemical radical functionalization of diazo compounds via direct anodic oxidation of diazo compounds should be feasible by using suitable nucleophiles. Herein, we report an electrochemical bromination reaction of diazo compounds via convergent paired electrolysis with CBr_4_ as a bromine source and CH_2_Cl_2_ as a hydrogen source (Schemeb). The diazo compounds undergo anodic oxidation to generate carbon radical cations. Meanwhile, CBr_4_ is reduced at the cathode to produce the final product. This work is the first example of the direct anodic oxidation of diazo compounds to couple with the nucleophile. This paired electrolysis mode differs from the previous electrochemical reactions of diazo compounds, which involve only a single electrode in the reaction. Moreover, this reaction provides a new and efficient method for the synthesis of α-bromo phosphonates.
To test this hypothesis, there are two problems that should be taken into consideration. One is the high oxidative potential of diazo compounds, which makes the anodic oxidation difficult. Thus, the nucleophile may preferentially oxidize at the anode to form a radical species. The second challenge is whether the nucleophilic precursors could couple with the in situ-generated radical cation intermediate from diazo compounds in this electrosynthesis. Our proof-of-concept work commenced by testing the reaction of diethyl diazo(phenyl)methyl phosphonate (1a) with tetrabromomethane (CBr_4_, 2) as a bromine source under electrochemical conditions (Table). Extensive optimization revealed that the reaction of 1a with CBr_4_ could occur to generate desired α-bromo phosphonate 3a in 58% yield when it was conducted in dichloromethane with nBu_4_NBF_4_ as an electrolyte in an undivided cell equipped with a graphite anode and a graphite cathode under a constant current of 5 mA for 4 h at room temperature (entry 1). Then, several common organic solvents were screened for this electrochemical transformation. Acetonitrile and 1,2-dichloroethane were also suitable solvents for this reaction, achieving desired product 3a in 46 and 57% yields, respectively (entries 2 and 3). However, no desired product 3a was detected when the reaction used protic solvent MeOH as the reaction medium (entry 4). This is mainly due to the dual roles of the solvent, reaction media, and hydrogen atom source. Other electrolytes, including nBu_4_NI, nBu_4_NPF_6_, nBu_4_NAc, and nBu_4_NClO_4_, were also used for this reaction (entries 5–8). nBu_4_NPF_6_ was demonstrated to be the most effective one, which afforded a slightly increased yield (60%, entry 6). No desired product 3a was detected when the reaction was conducted by using nBu_4_NI or nBu_4_NAc as the electrolyte (entries 5 and 7). It was found that other electrode combinations, such as (+)C|Pt(−), (+)Pt|Pt(−), and (+)Pt|C(−), also worked well to furnish corresponding product 3a, and the best yield was observed using (+)C|Pt(−) as electrodes (65% yield, entry 9). Finally, variations in the loading amount of diazo compound 1a and current density were carried out (entries 12–15). Fortunately, the yield could be further increased to 96% when 3.0 equiv of diazo 1a was used (entry 13). The current density is crucial for this transformation, and decreased yields were observed under a decreased or increased current density (entries 14 and 15).
After establishing the optimized reaction conditions, we then examined the substrate scope of diazo compounds 1 for this electrochemical reaction to access structurally diverse α-bromo phosphonates (Scheme). Generally, all of the employed aryl diazos bearing substituents at different positions of the phenyl ring, including alkyl, halo, and aryl, were all tolerated in this electrochemical bromination reaction, providing desired α-bromo phosphonates 3 in good to excellent chemical yields. Notably, the substrate with electron-withdrawing properties (CF_3_, 1m) worked well, leading to corresponding product 3m in 91% yield. However, no desired product was detected for the substrate with a strong electron-donating group (OMe, 1k). The reaction did not show an obvious steric hindrance effect. For example, ortho-, meta-, and para-methyl-substituted phenyl diazo substrates were all well engaged in the reaction, generating corresponding products 3b–3d in 73–74% yields. Subsequently, phenyl diazo substrates bearing functional groups were examined in this electrochemical reaction. We were delighted to find that phenyl diazos bearing a cyano group (1n) and an ester group (1p) were also compatible with this transformation, affording desired products 3n and 3p in 87 and 70% yields, respectively. It is worth mentioning that naphthyl-substituted diazo was also a viable substrate in this reaction, affording corresponding product 3q in 69% yield. Finally, various phosphonate groups on the diazo substrates were examined. Pleasingly, substrates containing different esters, including methyl, isopropyl, and butyl, also displayed good reactivity to provide products 3r–3t in 57–86% yields. The length of the alkyl chain shows an effect on the reaction outcome, and a 57% yield was obtained for the case with the butyl phosphonate group (3t). To demonstrate the robust nature of the protocol, we performed the reaction on a 2.0 mmol scale, and desired product 3a was obtained in 81% yield, underlining the promising potential of this electrochemical process to construct functionalized α-bromo phosphonates.
Encouraged by the above results, we further examined the reaction generality by employing diazo compounds bearing different substituents as substrates (Scheme). First, the stable diazo ester (1aa) was used as a substrate in this electrochemical transformation under the standard conditions (Schemea), which did not afford desired bromination product 4. Then, the reaction of the α-diazo ester bearing a phenyl group (1ab) with CBr_4_ was carried out. Interestingly, α-diazo-α-phenyl ester (1ab) was compatible with the procedure to generate desired product 5 in 57% yield. After careful isolation of the reaction mixture, α-keto ester 6 was also obtained in 10% yield, which was generated from the reaction of diazo with oxygen.? It is worth mentioning that introducing a phenylcarbonyl group onto the diazo phosphonate (1ac) was also successful, and the reaction generated desired α-bromo-β-keto phosphonate 7 in 23% yield. Finally, β-diazo-α,α-difluoroethylphosphonate (1ad) was also assayed in this electrochemical reaction. No desired bromination product was detected with all of the diazo 1ad consumed, and ketone product 8 was obtained in 28% yield.
To determine whether the diazo substrates are oxidized at the anode, several control experiments, as well as the cyclic voltammetry experiments, were carried out. First, a reaction between diazo phosphonate 1a and CBr_4_ 2 was conducted under the standard conditions but without passing any electricity (Schemea). The corresponding bromination of diazo 3a was not observed, with almost all of the starting materials remaining. Then, a radical-trapping reaction of diazo 1a and CBr_4_ 2 was carried out with the addition of 10.0 equiv of radical scavenger 2,2,6,6-tetramethyl-1-piperidinyl-N-oxyl (TEMPO). The reaction was totally suppressed, and no desired product 3a was obtained (Schemeb). TEMPO-trapped dichloromethyl adduct 9 was detected by ESI-MS. These results clearly reveal that this electrochemical transformation involves a dichloromethyl radical species. Then, radical trapping experiments with the addition of 10.0 equiv of 1,1-diphenylethene (DPE) or butylated hydroxytoluene (BHT) were conducted. The formation of desired bromination product 3a was inhibited in these two reactions. Fortunately, DPE-trapped tribromomethyl radical 10 was detected by ESI-MS, which indicates that the tribromomethyl radical may be generated in this electrochemical process. Also, radical intermediate 11 generated from the bromination of diazo substrate 1a was detected, and the result discloses that the α-bromo phosphonate radical is involved in this electrochemical process.
Subsequently, to gain information about the oxidation and reduction potentials of substrates and reagents in this electrochemical process, cyclic voltammetry experiments (CV) were conducted to elucidate the possible mechanism, and the results are shown in Figure. As shown in Figurea, there was an oxidation peak of diazo 1a observed at 2.0 V (vs Ag/AgCl), whereas no oxidation peak was observed for CBr_4_. On the other hand, there was a reductive peak at – 1.7 V (vs Ag/AgCl), while there were no reduction peaks observed for diazo 1a (Figureb). These results disclose that this reaction should involve the anodic oxidation of diazo 1a and the cathodic reduction of CBr_4_.?
Based on the above experimental results and related literature reports, ?−? ? ? ?,? combined with B3LYP-D3/6-311+G(d,p) density functional theory (DFT) calculations applying the SMD model to mimic the use of CH_2_Cl_2_ as a solvent,? a plausible reaction mechanism was proposed in Scheme. Initially, diazo substrate 1a undergoes anodic oxidation at the anode to generate radical cation intermediate A with the release of N_2_. This process proceeds via a transition state with an energy barrier of 28.3 kcal/mol, delivering radical cation intermediate A in an endergonic step (ΔG = 11.0 kcal/mol). On the other hand, the cationic reduction of CBr_4_ occurs to generate a bromine anion and CBr_3_ radical.? The coupling reaction of radical cation intermediate A with a bromine anion gives radical intermediate B in a highly exergonic step (ΔG = – 59.0 kcal/mol). In a subsequent step, generated radical intermediate B absorbs the hydrogen atom from dichloromethane to generate desired product 3a (pathway a) and forms the dichloromethyl radical, which was detected by ESI-MS. The Gibbs energy associated with this final step was found to be 8.8 kcal/mol. In an alternative pathway (pathway b), radical cation intermediate A may absorb the hydrogen atom from dichloromethane, leading to the formation of cation intermediate C. According to DFT calculations, the formation of cationic intermediate C was found to proceed with an exergonicity of −10.7 kcal/mol. Then, intermediate C undergoes nucleophilic attack by a bromine anion to afford desired product 3a (pathway b) with an exergonicity of −41.4 kcal/mol. DFT calculations suggest that a radical cation can form without immediate N_2_ release, but this pathway has a higher barrier (ΔG ^⧧^ = 34.3 kcal/mol) than that of the prompt N_2_ extrusion route (ΔG ^⧧^ = 28.3 kcal/mol), making it less favorable.
We also investigated by means of DFT calculations the reactivity of several diazo compounds bearing different substituents by analyzing the energy barrier associated with the transition state, which delivers radical cation intermediate A (Figure). As mentioned above, the activation Gibbs energy for the transition state of diazo phosponate 1a was computed to be 28.3 kcal/mol. In contrast, for diazo ester (1aa) a Gibbs energy barrier of 37.5 kcal/mol was computed, which is in line with the experimental observation of no formation of desired bromination product 4. However, for α-diazo-α-phenyl ester (1ab), the barrier was lower (ΔG ^⧧^ = 28.8 kcal/mol), showing the feasibility for the reaction under study, in agreement with the obtained 57% yield of 5. Interestingly, when introducing a phenylketo group to the diazo phosphonate (1ac) which experimentally afforded desired product 7 with 23% yield, it was found to have an activation Gibbs energy of 33.0 kcal/mol. Finally, for fluorinated diazo compound β-diazo-α,α-difluoroethylphosphonate (1ad), a barrier of 29.0 kcal/mol was computed.
In summary, we have developed for the first time an electrochemical bromination reaction of diazo compounds via convergent paired electrolysis, affording α-bromo phosphonates as products in up to 96% yield. Combined experimental and computational studies disclose that the reaction proceeds via direct anodic oxidation to generate carbon radical cations to couple with the bromide anion generated at the cathode by using CH_2_Cl_2_ as a hydrogen source. This reaction represents a new electrochemical mode of diazo compounds and affords a sustainable and practical strategy for the synthesis of α-bromo phosphonates.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Nawrat C. C.Moody C. J.Natural products containing a diazo group Nat. Prod. Rep.2011281426144410.1039/c 1np 00031 d 21589994 · doi ↗ · pubmed ↗
- 2Mix K. A.Aronoff M. R.Raines R. T.Diazo Compounds: Versatile Tools for Chemical Biology ACS Chem. Biol.2016113233324410.1021/acschembio.6b 0081027739661 PMC 5161546 · doi ↗ · pubmed ↗
- 3a Cheng Q.Deng Y.Lankelma M.Doyle M. P.Cycloaddition Reactions of Enol-Diazo Compounds Chem. Soc. Rev.2017465425544310.1039/C 7CS 00324 B 28726896 PMC 5575991 · doi ↗ · pubmed ↗
- 4a Zhang Z.Gevorgyan V.Visible Light-Induced Reactions of Diazo Compounds and Their Precursors Chem. Rev.20241247214726110.1021/acs.chemrev.3c 0086938754038 PMC 12503537 · doi ↗ · pubmed ↗
- 5Khanal H. D.Thombal R. S.Maezono S. M. B.Lee Y. R.Designs and Strategies for the Halo-Functionalization of Diazo Compounds Adv. Synth. Catal.20183603185321210.1002/adsc.201800340 · doi ↗
- 6a Zeng L.Wang J.Wang D.Yi H.Lei A.Comprehensive Comparisons between Directing and Alternating Current Electrolysis in Organic Synthesis Angew. Chem., Int. Ed.202362 e 20230962010.1002/anie.20230962037606535 · doi ↗ · pubmed ↗
- 7He Z.Zhao W.Li Y.Yu Y.Huang F.Electrochemical S–H and O–H insertion reactions from thiols or salicylic acids with diazo esters Org. Biomol. Chem.2022208078808210.1039/D 2OB 01273 A 36200479 · doi ↗ · pubmed ↗
- 8Yang D.Guan Z.Peng Y.Zhu S.Wang P.Huang Z.Alhumade H.Gu D.Yi H.Lei A.Electrochemical oxidative difunctionalization of diazo compounds with two different nucleophiles Nature Commun.202314147610.1038/s 41467-023-37032-836928311 PMC 10020561 · doi ↗ · pubmed ↗