Molecular Prevalence and Genotypic Diversity of Theileria equi in Xinjiang, China, Based on Three Genes
Sinan Qin, Telieke Kulabieke, Duman Mizhamuhan, Mengyuan Zhang, Min Jin, Gulibositan Abula, Mengjie Pi, Haorui Wang, Yang Zhang, Qingyong Guo

TL;DR
This study examines the spread and genetic diversity of Theileria equi, a tick-borne parasite affecting horses in Xinjiang, China, using three genes to identify regional differences and dominant genotypes.
Contribution
The first comprehensive genotyping of Theileria equi in Xinjiang using three molecular markers and haplotype network analysis.
Findings
The overall infection rate of Theileria equi in Xinjiang was 38.41%, with Tacheng having the highest prevalence at 86.27%.
Genotype E of the 18S rRNA gene and genotype A of the EMA-1 gene were the most prevalent in the region.
Urumqi showed the highest genetic diversity of Theileria equi, suggesting it may be a hotspot for parasite evolution.
Abstract
Equine theileriosis, a disease caused by the protozoan parasite Theileria equi and trans-mitted by ticks, represents a significant threat to equine health and the equine industry in Xinjiang, China. To investigate the current prevalence and genetic characteristics of the parasite, we conducted a molecular survey and genetic analysis. Blood samples were collected from 440 apparently healthy horses across four regions (Altay, Ili, Tacheng, and Urumqi). The overall infection rate was 38.41%, with prevalence varying significantly by region; it was highest in Tacheng (86.27%) and lowest in Altay (20.88%). Genetic characterization based on three target genes revealed the following: analysis of the 18S rRNA gene identified two distinct genotypes (E and A), with genotype E being overwhelmingly dominant. All parasites tested belonged to genotype A for the EMA-1 gene. For the mitochondrial COI…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
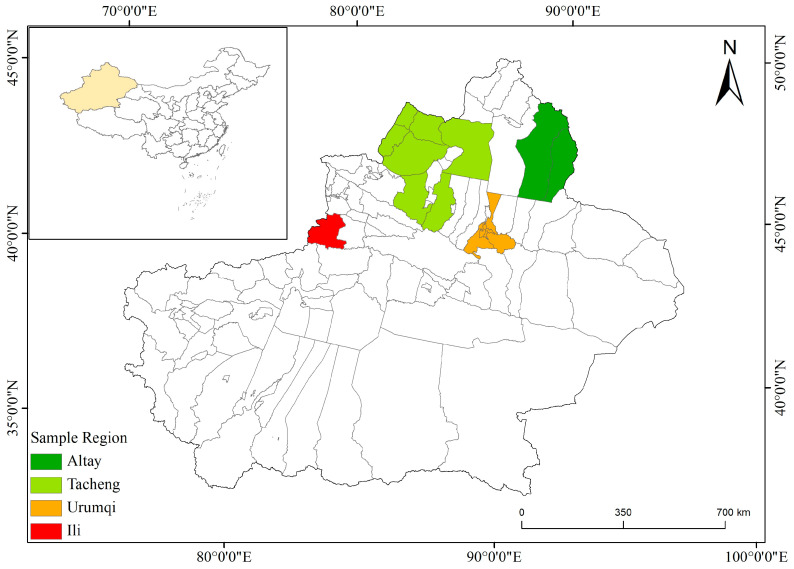 Figure 1
Figure 1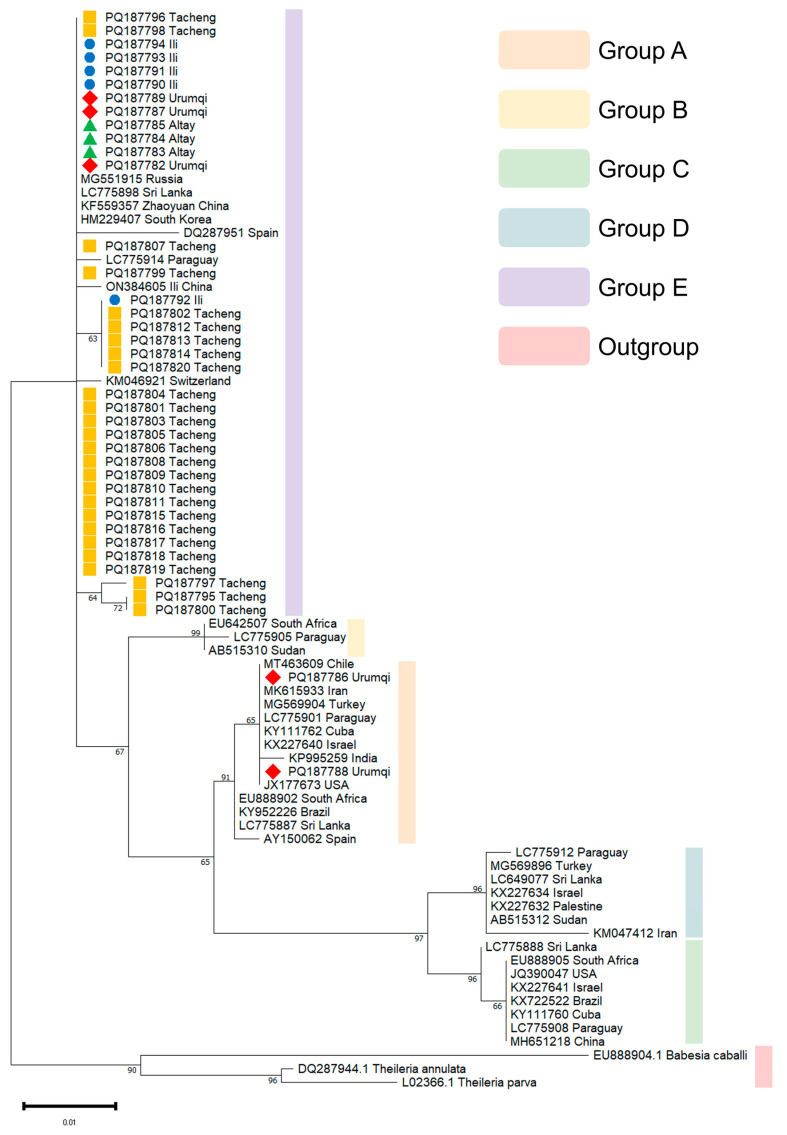 Figure 2
Figure 2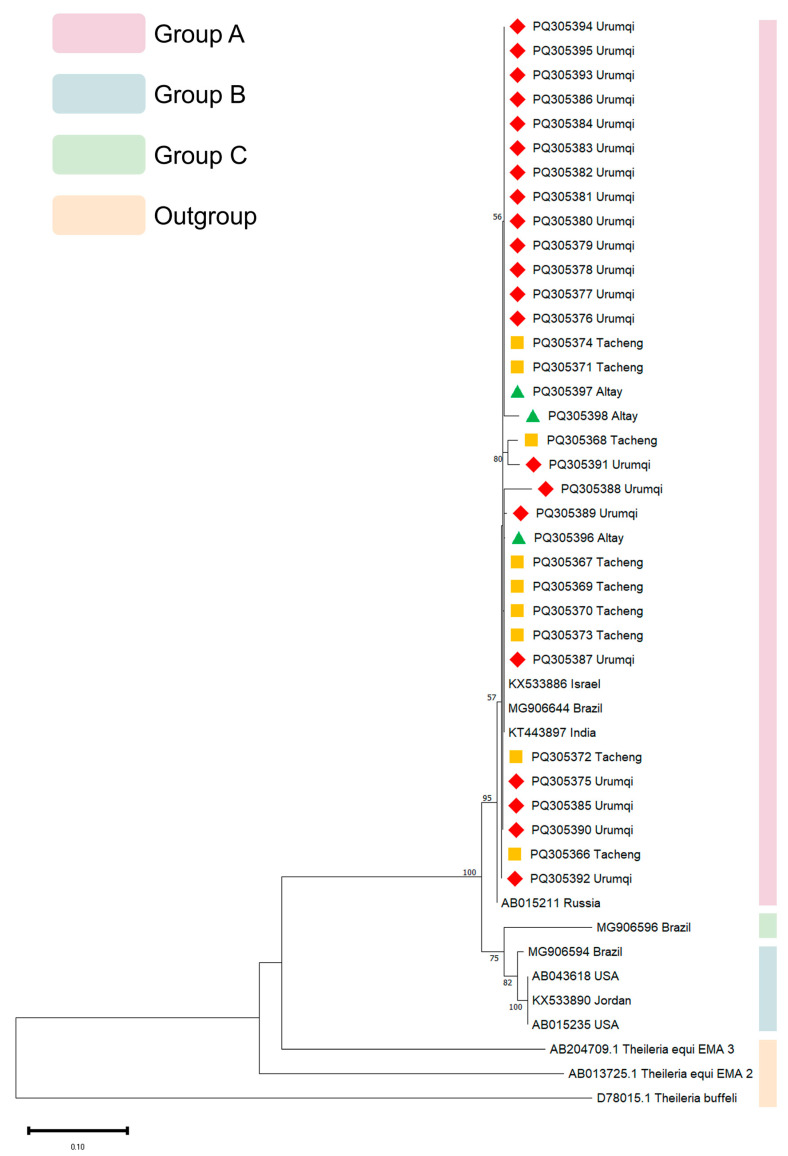 Figure 3
Figure 3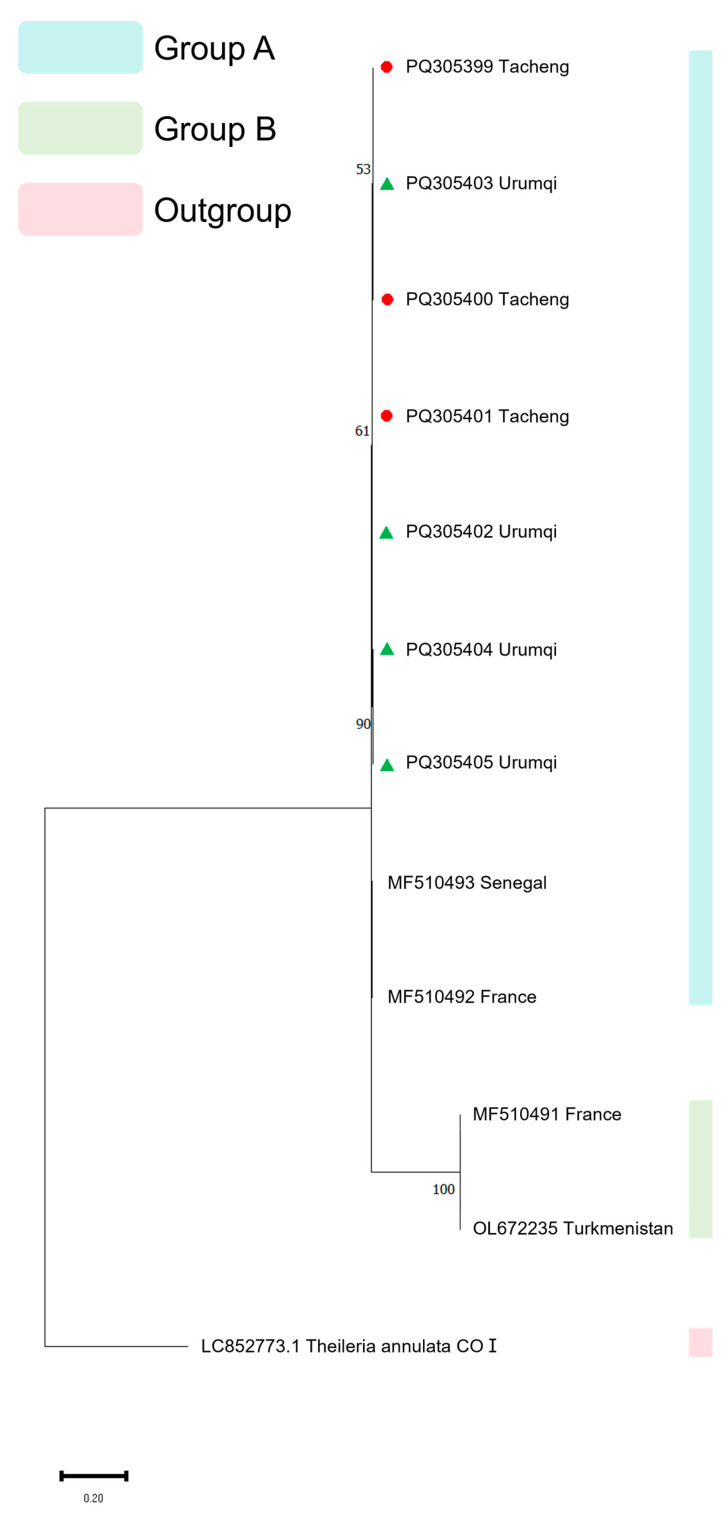 Figure 4
Figure 4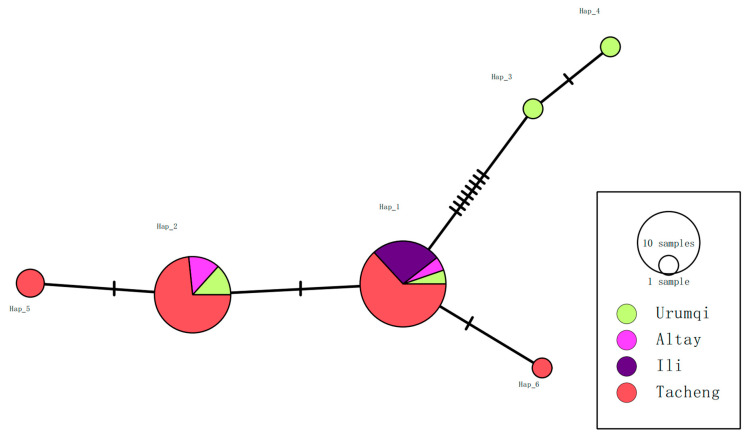 Figure 5
Figure 5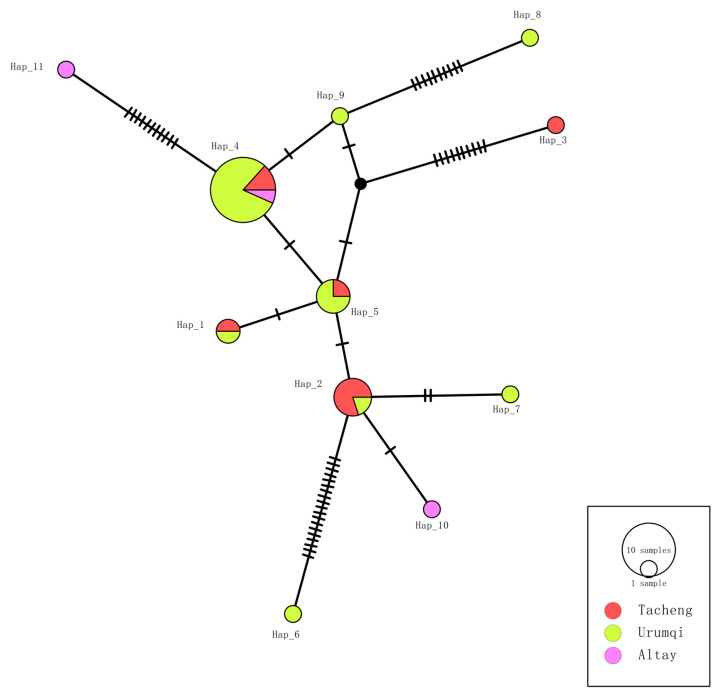 Figure 6
Figure 6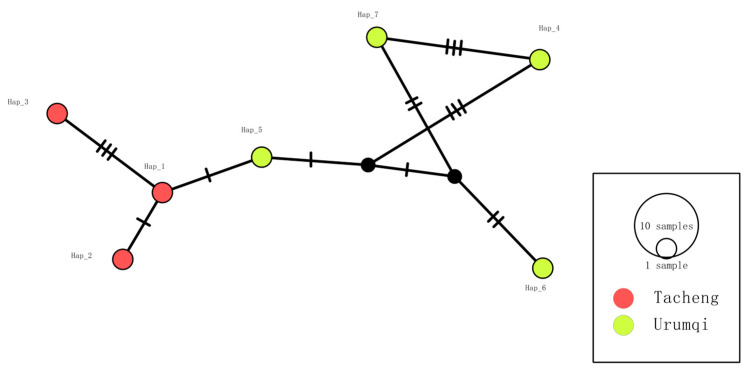 Figure 7
Figure 7- —Special Projects of the Central Government in Guidance of Local Science and Technology Development
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsVector-borne infectious diseases · Parasitic Diseases Research and Treatment · Parasites and Host Interactions
1. Introduction
Equine piroplasmosis is a tick-borne disease that poses a significant economic burden on the equine industry worldwide. Except for a few countries, such as Japan, the United States, and Australia, where it has not been reported, the disease is prevalent in many other countries [1]. Equine piroplasmosis is caused by three pathogens: Theileria equi (formerly described as Babesia equi), B. caballi, and T. haneyi [2,3,4,5,6].
These parasites infect all equid species, including horses, donkeys, mules, and zebras; however, clinical disease is rare in donkeys, mules, and zebras, allowing the disease to spread more easily and posing a global threat to equine health and productivity [2,3,7,8,9]. Infection with T. equi and B. caballi produces similar clinical signs, including fever, inappetence, icterus, and, in some severe cases, even death; however, clinical presentation tends to be more severe in cases of T. equi infection [2,10,11,12]. In contrast, the recently identified T. haneyi is rarely associated with clinical signs, even in splenectomized horses [12,13]. Consequently, equine piroplasmosis not only reduces the production performance of equine animals but also leads to increased veterinary costs, expenses for tick control, and restrictions on animal movement [2,14]. As an example, the joint APHIS-VS and Florida B. caballi eradication program took 25 years and cost USD 12 million for tick inspections, testing, the treatment of infected horses, and transportation restrictions [15].
In endemic regions, most infected horses are asymptomatic; however, these animals remain infectious, underscoring the importance of surveillance [14,16]. Since chronic carriers do not exhibit clinical symptoms but still retain infectivity, monitoring them in endemic areas is crucial for assessing the transmission risk of equine piroplasms, especially T. equi [14,17]. Generally, in Xinjiang, the T. equi infection rate is higher than that of B. caballi, and infections with T. equi typically persist for life [2,10,11,12,18,19,20]. Xinjiang is one of China’s most important livestock regions and has a rapidly developing horse industry. The total horse inventory in Xinjiang in 2020 was 954,500, meaning Xinjiang had the most horses in China; this number is steadily rising. Xinjiang is also a hyperendemic area for T. equi. The T. equi infection rate in some areas of Xinjiang was 40.8% in 2014, 39.5% in 2018, and 23.8% in 2021 [19,21,22]. Therefore, continuous monitoring and effective control measures in Xinjiang are essential to manage the T. equi transmission risk.
The taxonomic classification of T. equi has been controversial since its discovery [2,23,24]. Although the pathogen was ultimately named T. equi, additional data are still needed to determine its final classification. Genotype differences not only affect the diagnostic results but also lead to different clinical manifestations and treatment outcomes [25,26,27,28,29].
The 18S rRNA gene was employed as the primary marker for species-specific detection and broad-range genotyping, as it represents the most widely used genetic marker for phylogenetic analysis of piroplasms due to its conserved regions [30]. EMA-1, a major merozoite surface protein of T. equi that plays a crucial role in parasite recognition, adhesion, and invasion processes. Its immunogenic properties make it a promising vaccine candidate antigen, and a competitive ELISA based on EMA-1 has been widely adopted for serological diagnosis. Furthermore, EMA-1 serves as an excellent target for investigating intraspecific genetic variation within T. equi [7,23,29,31]. Mitochondria, as the core organelles for energy metabolism in eukaryotes, possess mitochondrial genomes that serve as ideal molecular markers for phylogenetic reconstruction due to their moderate evolutionary rate and lack of recombination, meaning that they can play a pivotal role in phylogenetic studies of apicomplexan parasites [32,33]. These mitochondrial genomes not only help resolve taxonomic controversies but also reveal cryptic diversity and speciation events [34]. Furthermore, mitochondrial functional research is equally critical for identifying potential drug targets and investigating resistance mechanisms [34,35]. The mitochondrial genome of T. equi exhibits a unique structural feature, demonstrating near-complete loss of synteny compared to species like B. bigemina, B. caballi, B. gibsoni, and T. orientali [36]. However, current phylogenetic studies on T. equi based on the mitochondrial genome remain limited [12], which significantly hindering population genetics research and the optimization of T. equi control strategies.
Some studies have demonstrated that the 18S rRNA genotype A of T. equi isolates is more likely to induce clinical symptoms in infected horses [29,37]. Moreover, there are also some studies indicating that imidocarb dipropionate can eliminate infections caused by genotype A of T. equi, but not those caused by T. haneyi [25,27]. Variations in antigen expression or treatment sensitivity among different T. equi genotypes could lead to changes in accuracy or sensitivity during diagnosis [4,38]. However, research on T. equi genotypes has mainly focused on the 18S rRNA and EMA-1 genes, while there is relatively less research on mitochondrial genotypes [12]. Therefore, the objectives of the present study were to (1) investigate the prevalence of T. equi during the period 2023–2024, (2) further characterize the genotypic composition of this pathogen, and (3) analyze its haplotype diversity.
2. Materials and Methods
2.1. Blood Sample Collection and DNA Extraction
The sample collection began in May 2023 and finished in May 2024. A total of 440 equine blood samples were collected in Xinjiang, China. The study area has a temperate continental arid and semi-arid climate. The total sample size was calculated to estimate the prevalence of T. equi in Xinjiang with a 95% confidence level and a 5% margin of error, based on a previously reported prevalence of 38.9% in China [18]. None of the horses exhibited clinical symptoms at the time of sampling.
Of the samples, 249 were collected in the Altay region, 88 were collected in the Ili region, 51 were collected in the Tacheng region, and 52 were collected in Urumqi (Figure 1). Approximately 5 mL of blood was collected from the external jugular vein of each horse into an EDTA-coated vacutainer tube. DNA was extracted using the TIANamp Blood DNA kit (TIANGEN, Beijing, China) according to the manufacturer’s instructions, and then stored at −20 °C until used. All animal protocols were reviewed by the Institutional Animal Care and Use Committee of Xinjiang Agricultural University (protocol number: GB/T 35892-2018) [39].
2.2. PCR Detection of T. equi
All 440 DNA samples underwent screening using T. equi-specific PCR assays. Briefly, the PCR assay for detecting T. equi was conducted with forward (5′-TCGAAGACGATCAGATACCGTCG-3′) and reverse primers (5′-TGCCTTAAACTTCCTTGCGAT-3′) targeting the 18S ribosomal RNA gene, which produce a 392 bp product [40]. The PCR was performed in a 25 μL mixture consisting of 2 × Taq PCR MasterMix II (CoWin, Taizhou, China), 2 μL of template DNA, 5 pmol of each primer, and 6.5 μL DNase/RNase-free water. The mixture was heated for 10 min at 96 °C, followed by 40 cycles of 1 min at 96 °C, 1 min annealing at 60.5 °C, and 1 min extension at 72 °C, before a final extension for 10 min at 72 °C. The success of amplification was verified by electrophoresis on a 1% agarose gel.
2.3. PCR Detection and Sequencing of T. equi Genotypes
Samples positive for T. equi were further used to detect the 18S rRNA, EMA-1, and COI genes. The PCR mixture was the same as that previously mentioned. The primer sequences and annealing temperature are shown in Table 1 [5,41,42]. The success of amplification was verified by electrophoresis on a 1% agarose gel. Positive PCR products were sequenced using Sanger technology (Sangon Biotech, Shanghai, China). The results obtained after sequencing were analyzed using the NCBI BLAST+2.17.0 algorithm, and the sequence data generated were submitted to GenBank under the following accession numbers: 18S rRNA: PQ187782-PQ187820, EMA-1: PQ305366-PQ305398, COI: PQ305399-PQ305405.
2.4. Phylogenetic Analysis Based on Multi-Gene Sequences
Three phylogenetic trees were generated utilizing 18S rRNA, EMA-1, and COI gene sequences. For the 18S rRNA gene PCR products, 39 samples yielding clear and distinct bands were selected for sequencing; for the EMA-1 gene PCR products, 33 such samples were chosen for sequencing; and for the COI gene PCR products, 7 samples with a single clear and distinct band were selected for sequencing. Of the 39 samples successfully sequenced for the 18S rRNA gene, 26 were from Tacheng (PQ187795-PQ187820), 5 were from Ili (PQ187790-PQ187794), 5 were from Urumqi (PQ187782, PQ187786-PQ187789), and 3 were from Altay (PQ187783-PQ187785). For the EMA-1 gene, of the 33 samples successfully sequenced, 9 were from Tacheng (PQ305366-PQ305374), 21 were from Urumqi (PQ305375-PQ305395), and 3 were from Altay (PQ305396-PQ305398). Only 7 samples were successfully sequenced for the COI gene, 3 from Tacheng (PQ305399-PQ305401) and 4 from Urumqi (PQ305402-PQ305405). Multiple sequence alignment of the obtained sequences was generated using Clustal W within MEGA 11 software, using a gap opening penalty of 15 and gap extension penalty of 6.66 for the pairwise and multiple alignments, respectively.
The Compute Nucleotide Composition function in MEGA 11 [43] was used to determine the nucleotide composition of the sequence. DnaSP 6 software [44] was used to calculate the number of haplotypes, haplotype diversity, and nucleotide diversity. Meanwhile, the haplotype network was constructed using PopART 1.7 software [45].
The Maximum Likelihood (ML) approach and the Kimura 2-parameter (K2P) model with 1000 bootstrap samplings were used to infer the evolutionary history using MEGA 11. In the construction of the 18S rRNA phylogenetic tree, the 18S rRNA gene sequences of T. annulata (DQ287944.1), T. parva (L02366.1), and B. caballi (EU888904.1) were used as the outgroup. In the construction of the EMA-1 phylogenetic tree, the EMA-2 (AB013725.1) and EMA-3 (AB204709.1) genes of T. equi, as well as major piroplasm surface protein gene of T. buffeli (D78015.1), were used as the outgroup. In the construction of the COI phylogenetic tree, the COI (LC852773.1) of T. annulata, was used as the outgroup.
2.5. Statistical Analyses
The 95% confidence intervals (CIs) for prevalence rates were calculated using VassarStats (VassarStats: Statistical Computation Web Site) to estimate the potential ranges of true prevalence. Statistical significance (p values) was determined with SPSS 27 (SPSS Software, IBM, Armonk, NY, USA) [46]. To determine if the differences in infection rates among the four sampling regions (Altay, Ili, Tacheng, and Urumqi) were statistically significant, a Pearson’s chi-squared (χ^2^) test of independence was performed. p-values of less than 0.05 were considered to indicate statistical significance.
3. Results
3.1. Molecular Detection of T. equi
Among the 440 sampled horses, 169 were positive for T. equi, corresponding to an overall prevalence of 38.41% (95% CI: 33.87–43.15). Regionally, the prevalence varied significantly: 52 (20.88%), 44 (50.00%), 44 (86.27%), and 29 (55.77%) positive cases were detected in Altay, Ili, Tacheng, and Urumqi, respectively. Using the chi-squared test, we found statistically significant differences in infection rates between regions (p < 0.001). Further comparing the infection rates in different regions, we found that, except for the infection rates in Ili and Urumqi, which had no statistical differences (p = 0.461), the pairwise comparisons between other regions were statistically different (p < 0.001) (Table 2).
3.2. Phylogenetic Analyses
For 18S rRNA genotyping, two distinct genotypes (A and E) were identified among the 39 sequenced samples. Thirty-seven (94.9%) sequences clustered within genotype E, while the remaining two (5.1%) sequences formed a separate clade representing genotype A. Within the genotype E clade, our sequences showed close phylogenetic affinity with T. equi isolates collected in diverse geographic regions, including Russia (MG551915), Sri Lanka (LC775898), South Korea (HM229407), Spain (DQ287951), and Paraguay. The two genotype A sequences from Urumqi clustered closely with reference strains from Chile (MT463609), Iran (MK615933), Turkey (MG569904), Paraguay (LC775901), Cuba (KY111762), Israel (KX227640), India (KP995259), United States (JX177673), South Africa (EU888902), Brazil (KY952226), Sri Lanka (LC775887), and Spain (AY150062).
Geographically, all samples from Tacheng, Ili, and Altay belonged to genotype E, whereas Urumqi exhibited co-circulation of both genotypes, A (PQ187786, PQ187788) and E (PQ187789, PQ187787, PQ187782) (Figure 2).
All EMA-1 gene sequences generated in this study clustered within genotype A, demonstrating close phylogenetic affinity to previously reported strains from Israel (KX533886), Brazil (MG906644), India (KT443897), and Russia (AB015211). Genotype B comprised a single sequence from Brazil (MG906596), while genotype C included two sequences from the USA (AB043618, AB015235), one from Brazil (MG906594), and one from Jordan (KX533890) (Figure 3).
The phylogenetic tree analysis of the COI gene revealed that all seven sequenced samples clustered together in a single clade, exhibiting a closer evolutionary relationship with isolates from Senegal and France. Concurrently, another French isolate and a Turkmenistan isolate formed a distinct, independent clade (Figure 4).
3.3. Haplotype Analyses
3.3.1. 18S rRNA Gene
Among the 39 sequenced samples, six haplotypes were identified for the 18S rRNA gene. Tacheng and Urumqi exhibited the highest haplotype richness (n = 4 each), while Altay and Ili had lower richness (n = 2 and n = 1, respectively). Haplotype diversity (Hd) varied markedly across regions: Ili had the lowest Hd (0.0000), reflecting a single fixed haplotype, while Urumqi displayed the highest Hd (0.9000). Nucleotide diversity (π) followed a similar trend and was highest in Urumqi (0.01747) (Table 3). Notably, Hap_1 was shared across all four regions, indicating a widespread ancestral lineage, whereas Hap_2 was restricted to Urumqi, Altay, and Tacheng (Figure 5).
3.3.2. EMA-1 Gene
For the EMA-1 gene, 11 haplotypes were identified among 33 samples. Urumqi had the highest number of haplotypes (n = 8), Altay exhibited the greatest haplotype diversity (Hd = 1.000) due to its smaller sample size (n = 3), and Urumqi showed the lowest diversity (Hd = 0.671). Nucleotide diversity was also highest in Altay (π = 0.01688) (Table 3), suggesting localized genetic drift or selection pressure. Haplotype sharing patterns revealed Hap_4 as unique to three regions, whereas Hap_1, Hap_2, and Hap_5 were shared between Tacheng and Urumqi (Figure 6).
3.3.3. COI Gene
COI gene analysis of the seven sequenced samples identified seven distinct haplotypes, with Tacheng and Urumqi harboring three and four haplotypes, respectively. Both regions showed high haplotype diversity (Hd = 1.0000), but nucleotide diversity was relatively low in Tacheng (π = 0.00344) (Table 3), indicating a recent population expansion or purifying selection. The haplotype network revealed that there were no shared haplotypes between Tacheng and Urumqi (Figure 7), suggesting limited gene flow or ecological isolation.
4. Discussion
This molecular epidemiological study analyzed 440 equine samples from four regions in Xinjiang (Altay, Ili, Tacheng, and Urumqi) collected between 2023 and 2024. The overall prevalence of T. equi infection was 38.41% (95% CI: 33.87–43.15), consistent with a previous report from China (38.9%. 2018–2020) [18]. However, this infection rate is substantially higher than the global estimated prevalence of 29.4% [47]. Current data indicate that the prevalence of T. equi infection in China is notably higher than in some Asian countries (1.3% in Thailand [48]) and similar to that in others (24.8% in Kyrgyzstan [14] and 20.79% in India [7]), but lower than the very high rates reported in Sri Lanka (85.6%) [49] and Mongolia (78.2%) [38]. The extremely high prevalence of infection in Sri Lanka and Mongolia may be attributed to ecological and husbandry factors that are highly favorable to the tick-parasite cycle, such as the optimal climatic conditions for vector ticks, extensive pastoral management systems with limited acaricide use, and potentially high densities of equine hosts. The stable overall prevalence of T. equi infection reflects the characteristics of an endemic disease.
The results demonstrated geographical variations in the prevalence of T. equi across Xinjiang, potentially associated with temperature, humidity, and rainfall patterns [50] and the distribution of tick vectors. Known tick vectors of T. equi include Hyalomma excavatum, H. anatolicum, Rhipicephalus bursa, R. microplus, and Amblyomma cajennense, which have been reported in multiple regions worldwide [17]. In addition, studies in China confirm transmission by D. nuttalli, D. silvarum, D. niveus, and Rhipicephalus haemaphysaloides [51]. Tick species show distinct regional patterns. In Tacheng, the main tick species are H. asiaticum, H. marginatum, D. niveus, D. nuttalli, D. silvarum, D. marginatus, and Haemaphysalis punctata. The main tick species in Urumqi are H. asiaticum, H. anatolicum, and Ixodes persulcatus. In Ili, the main tick species are H. marginatum, H. dromedarii, D. silvarum, D. marginatus, H. asiaticum, H. punctata, R. turanicus, and I. persulcatus. In Altay, the main tick species are D. nuttalli, D. niveus, D. marginatus, D. silvarum, and H. punctata [52,53,54,55]. Therefore, we hypothesize that regional prevalence differences correlate with tick vector competence. For instance, the prevalence of infection in Urumqi (55.77%) being higher than that in Altay (20.88%) may stem from the presence of H. anatolicum, a highly efficient vector, despite this region hosting fewer tick species.
When compared with historical data, the prevalence of T. equi infection showed an upward trend across all surveyed regions. In Ili, the rate increased to 50.00% in the present study from earlier reports [21], while Tacheng showed a marked rise from 66.70% in 2020 to 86.27% in the current study [18]. Similarly, the prevalence of infection in Urumqi had surged from 17.7% (2018–2020) to 55.77% [56], while the prevalence in Altay had increased from 13.3% in 2016 to 20.88% in the current study [57].
Given the high prevalence of T. equi in Xinjiang, elucidating the characteristics of its genetic diversity is essential for formulating targeted control strategies. From an evolutionary perspective, low-pathogenicity genotypes may have a transmission advantage [29], as demonstrated in B. rossi genotype–pathogenicity correlations [58]. Genetic diversity influences diagnostic sensitivity [29] and the selection of vaccine antigens [31]. In this study, 18S rRNA genotype E was predominant (94.87%, 37/39), with only two genotype A cases identified (exclusively in Urumqi). The predominance of genotype E may reflect its Eurasian-wide distribution and asymptomatic phenotype, whereas genotype A is linked to clinical disease [12,50]. In China, genotypes A, B, C, and E of T. equi 18S rRNA gene have been reported. Genotype A is primarily distributed in Inner Mongolia, Tianjin, and Jilin; genotype B has been found in only one case in Jilin; genotype C is mainly distributed in Tianjin and Gansu; and E genotype has the broadest distribution, having been detected in multiple regions including Anhui, Hebei, Heilongjiang, Henan, Inner Mongolia, Jilin, Liaoning, Shandong, Sichuan, Tianjin, and Xinjiang [21,59,60,61]. In this study, we confirmed that genotype A is also present in Xinjiang, specifically in Urumqi, a region which harbors both A and E genotypes. This contrasts with Ili, Altay, and Tacheng, which contain genotype E exclusively. Notably, the genotype in the Ili region is consistent with our detection and sampling results from 2021 [21]. The study detected two genotypes (A and E) in Urumqi, whereas only a single genotype (E) was found in other regions. The genetic diversity of Urumqi may reflect its role as a transportation hub with frequent equine trade.
Despite extensive T. equi genotyping studies in China, most have focused on a single gene [18,20,59,61]. Notably, the combined utilization of 18S rRNA and EMA-1 genes can provide a more comprehensive solution for elucidating the genetic diversity of T. equi [7,29]. In this study, phylogenetic analysis of the EMA-1 gene revealed that all isolates belonged to genotype A, representing the first confirmation that the 18S rRNA genotype E/EMA-1 genotype A combinatorial pattern predominates among T. equi populations in Xinjiang.
Due to the limited availability of COI reference sequences for T. equi and the absence of a standardized genotyping system, we provisionally categorized the COI sequences of T. equi into two groups based on the phylogenetic tree results. The first group includes all Xinjiang isolates sequenced in this study, along with isolates from France and Senegal. The second group consists of another French isolate and an isolate from Turkmenistan. With further research, we anticipate that the genetic and evolutionary data for the COI gene in T. equi will be refined and expanded to establish a more comprehensive genotyping framework.
Haplotype diversity refers to the degree of genetic heterogeneity in combinations of single nucleotide polymorphisms (SNPs) exhibiting linkage disequilibrium within specific chromosomal regions and serves as a core indicator for evaluating genetic variation within populations [62]. In the field of parasitology, haplotype studies hold significant value for elucidating drug resistance mechanisms, guiding drug target screening, and optimizing prevention and control strategies [63]. For instance, specific haplotypes of Plasmodium species can enhance transmission efficiency by modulating the specific binding of gametocyte surface proteins to mosquito midgut receptors [64]. However, global research on T. equi haplotypes remains extremely limited [65], and there are no reports in China to date. To our knowledge, this study represents the first haplotype analysis of three genes in T. equi populations from Xinjiang, China.
Haplotype analysis of the 18S rRNA gene revealed six distinct haplotypes among 39 samples, with Tacheng and Urumqi exhibiting four haplotypes each. For the EMA-1 gene, haplotype analysis of 33 samples identified eight haplotypes in Urumqi. Similarly, COI gene haplotype analysis of seven samples detected four haplotypes in Urumqi. When considering the combined haplotype and nucleotide diversity results across all three genes, Urumqi demonstrated the highest levels for both parameters. This multi-locus enrichment of genetic diversity suggests that Urumqi may serve as an evolutionary hotspot for T. equi within the Xinjiang region. The distribution of Hap-1 in the 18S rRNA gene across all four regions and Hap-4 in the EMA-1 gene across three regions suggests that these two haplotypes may represent ancestral lineages. Alternatively, these haplotypes might carry advantageous mutations that have been independently selected for and retained across multiple regions due to their adaptive benefits.
Limitations: This study has several limitations. First, as detailed in the methods, individual demographic data (e.g., precise age, breed) and formal clinical assessments were not collected. Therefore, our analysis cannot identify host-specific risk factors for infection, and the reported prevalence represents the infection rate within a broadly defined, apparently healthy adult horse population. Future studies incorporating detailed host metadata would be valuable for a more comprehensive risk analysis. The analysis of the mitochondrial COI gene, while pioneering in the context of T. equi in China, was constrained by a small final sample size (n = 7). This limitation precludes strong conclusions regarding mitochondrial diversity and lineage structure. The technical difficulties associated with amplifying this locus from field-derived DNA are acknowledged. Nonetheless, these data provide a critical first step by confirming amplification feasibility and yielding initial sequences to populate international databases. The observed phylogenetic clustering with isolates from France and Senegal offers a testable hypothesis for future larger-scale studies. Establishing a comprehensive COI genotyping system remains a future priority, for which this study provides foundational data.
This study investigated the molecular prevalence of T. equi across four regions in Xinjiang, China, and elucidating the genetic characteristics of T. equi within the region. The results revealed varying levels of infection prevalence across all the investigated regions, with significant geographical variations potentially associated with the diversity of tick species in different areas. Genetic analyses demonstrated that 18S rRNA genotype E and EMA-1 genotype A were predominant among T. equi isolates in Xinjiang. Furthermore, this study performed the first haplotype analysis of T. equi in China, revealing Urumqi as a genetic diversity hotspot for the species.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Nugraha A.B. Cahyaningsih U. Amrozi A. Ridwan Y. Agungpriyono S. Taher D.M. Guswanto A. Gantuya S. Tayebwa D.S. Tuvshintulga B. Serological and molecular prevalence of equine piroplasmosis in Western Java, Indonesia Vet. Parasitol. Reg. Stud. Rep.2018141610.1016/j.vprsr.2018.07.00931014711 · doi ↗ · pubmed ↗
- 2Wise L. Kappmeyer L. Mealey R. Knowles D. Review of equine piroplasmosis J. Vet. Intern. Med.2013271334134610.1111/jvim.1216824033559 · doi ↗ · pubmed ↗
- 3Tamzali Y. Equine piroplasmosis: An updated review Equine Vet. Educ.20132559059810.1111/eve.12070 · doi ↗
- 4Knowles D.P. Kappmeyer L.S. Haney D. Herndon D.R. Fry L.M. Munro J.B. Sears K. Ueti M.W. Wise L.N. Silva M. Discovery of a novel species, Theileria haneyi n. sp., infective to equids, highlights exceptional genomic diversity within the genus Theileria: Implications for apicomplexan parasite surveillance Int. J. Parasitol.20184867969010.1016/j.ijpara.2018.03.01029885436 · doi ↗ · pubmed ↗
- 5Kalantari M. Sharifiyazdi H. Ghaemi M. Ghane M. Nazifi S. Theileria equi in the horses of Iran: Molecular detection, genetic diversity, and hematological findings Vet. Parasitol. Reg. Stud. Rep.20223610079210.1016/j.vprsr.2022.10079236436901 · doi ↗ · pubmed ↗
- 6Camacho A. Guitian F. Pallas E. Gestal J. Olmeda A. Habela M. Telford Iii S. Spielman A. Theileria (Babesia) equi and Babesia caballi infections in horses in Galicia, Spain Trop. Anim. Health Prod.20053729330210.1007/s 11250-005-5691-z 15934637 · doi ↗ · pubmed ↗
- 7Maharana B.R. Ganguly A. Potliya S. Kumar B. Singh H. Dash A. Khanna S. Molecular detection and characterization of prevailing Theileria equi genotype in equine from northern India Res. Vet. Sci.202417310527710.1016/j.rvsc.2024.10527738678846 · doi ↗ · pubmed ↗
- 8Knowles D.Jr. Kappmeyer L. Stiller D. Hennager S. Perryman L. Antibody to a recombinant merozoite protein epitope identifies horses infected with Babesia equi J. Clin. Microbiol.1992303122312610.1128/jcm.30.12.3122-3126.19921280648 PMC 270599 · doi ↗ · pubmed ↗