Cell Surface Vimentin Is an Attachment Factor That Facilitates Equine Arteritis Virus Infection In Vitro
Côme J. Thieulent, Sanjay Sarkar, Mariano Carossino, Mouli Bhowmik, Haining Zhu, Udeni B. R. Balasuriya

TL;DR
Researchers found that vimentin, a cell surface protein, helps the equine arteritis virus infect cells, even when the main receptor is absent.
Contribution
The study identifies vimentin as a novel attachment factor for equine arteritis virus infection in cells lacking the primary receptor.
Findings
A 57 kDa membrane protein identified as vimentin binds to equine arteritis virus.
Cells expressing vimentin are susceptible to equine arteritis virus infection.
Blocking vimentin or overexpressing it affects equine arteritis virus infection outcomes.
Abstract
Our laboratory identified the susceptible allelic variant of equine CXCL16 protein (EqCXCL16S) as an entry receptor for equine arteritis virus (EAV). However, EAV has a broad host cell tropism and infects cells that lack EqCXCL16S. Thus, we hypothesized that EAV interacts with other host cell protein(s) that facilitate EAV infection. A virus overlay protein-binding assay in combination with a Far-Western blot from EAV-susceptible equine pulmonary artery endothelial cells (EECs) and equine dermal fibroblasts (E. Derm) identified a 57 kDa protein, present in the membrane fraction of the protein lysate, as a possible EAV-binding protein. Subsequent LC-MS/MS analysis identified this 57 kDa protein as vimentin. Screening of different mammalian cell lines has shown that only cells expressing vimentin are susceptible to EAV infection. Pre-treatment of EECs with an anti-vimentin polyclonal…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
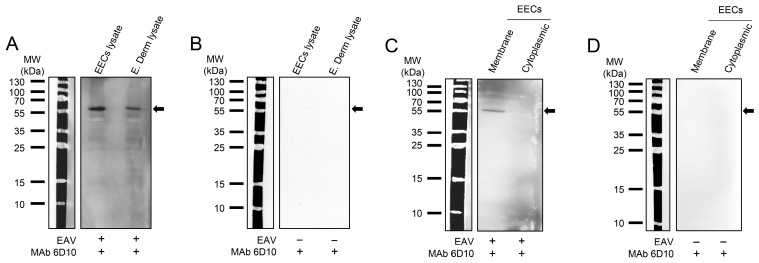 Figure 1
Figure 1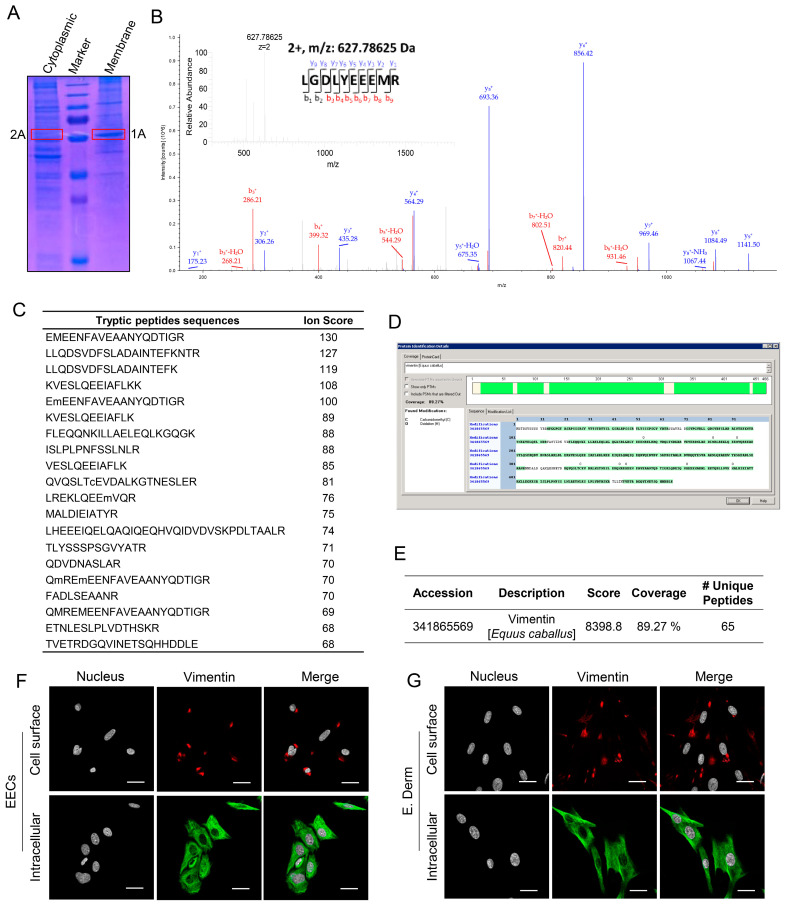 Figure 2
Figure 2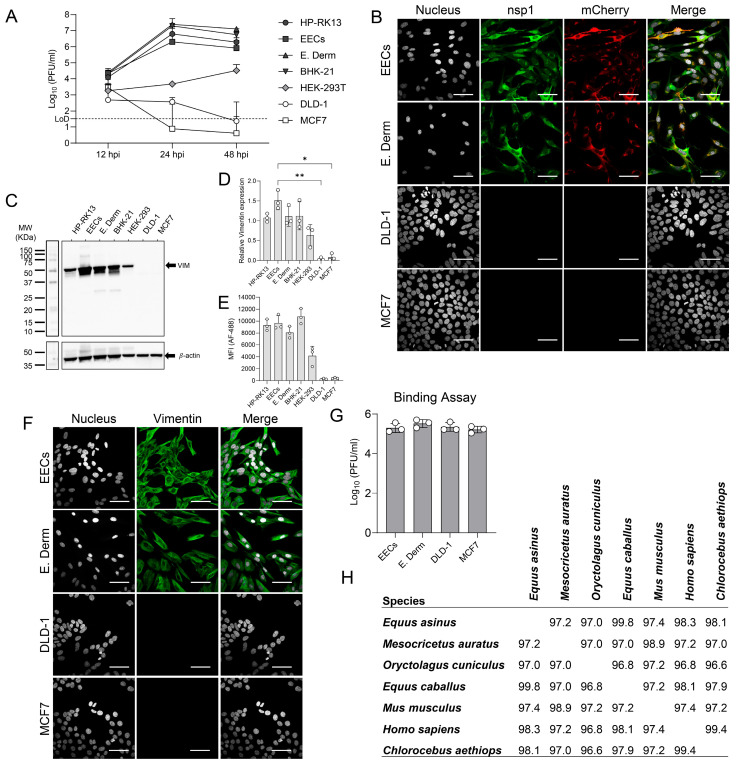 Figure 3
Figure 3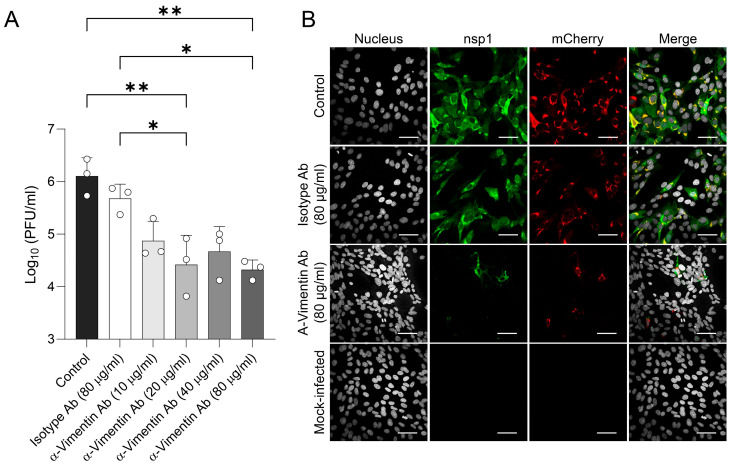 Figure 4
Figure 4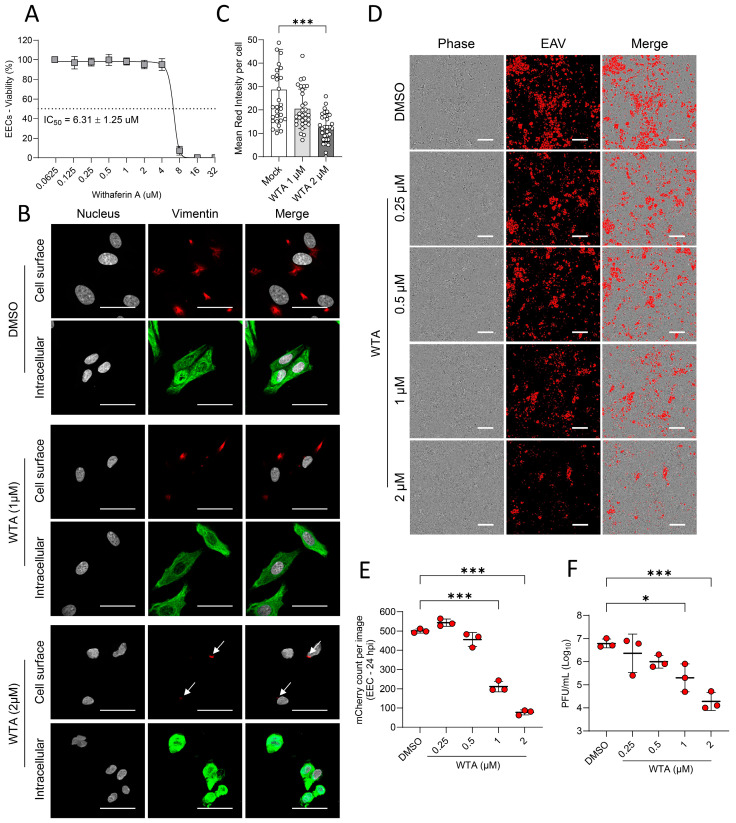 Figure 5
Figure 5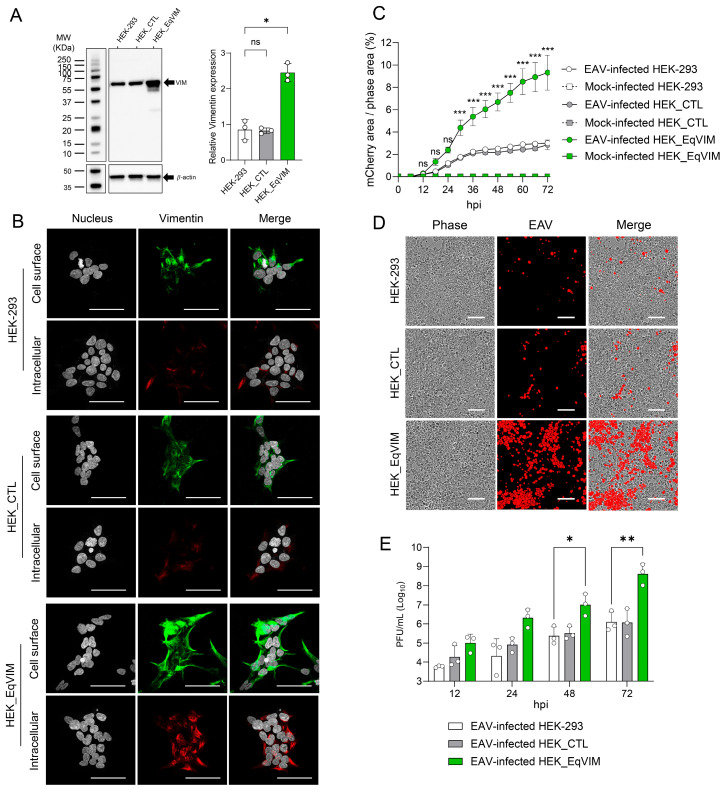 Figure 6
Figure 6- —USDA National Institute of Food and Agriculture to UBRB
- —self-generated funds
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAnimal Virus Infections Studies · Chemokine receptors and signaling · Herpesvirus Infections and Treatments
1. Introduction
Equine arteritis virus (EAV), recently renamed Alphaarterivirus equid, is a single-stranded, positive-sense RNA virus belonging to the genus Alphaarterivirus, subfamily Equarterivirinae, family Arteriviridae, order Nidovirales [1,2]. EAV was first isolated from the lung of a fetus during an abortion storm on a horse farm in Bucyrus, Ohio, in 1957 [3,4]. EAV is the causative agent of equine viral arteritis (EVA), a respiratory and reproductive disease with a worldwide prevalence, resulting in significant economic losses to the equine industry [5]. EAV infections are mostly subclinical, but they can be associated with a wide range of clinical signs, including respiratory distress, flu-like illness, dependent edema, conjunctivitis, periorbital or supraorbital edema, urticaria, and leukopenia [6,7,8,9,10,11,12,13]. Infection of pregnant mares may result in abortion or birth of congenitally infected foals, developing rapidly fatal bronchointerstitial pneumonia or pneumoenteric syndrome [14,15]. EAV is transmitted by the respiratory route, via infective aerosolized secretions, and by the venereal route, via infective semen derived from infected stallions [5]. Following infection, EAV establishes long-term persistent infection (LTPI) in the stallion’s reproductive tract, resulting in the continuous shedding of infectious virus in the semen [13,16]. Our previous studies demonstrated that EAV infects and persists in the ampullae of the stallion reproductive tract, exhibiting a specific tropism for vimentin-positive stromal cells within the lamina propria, as well as for CD8^+^ T cells and CD21^+^ B lymphocytes [13], despite the presence of strong serological, mucosal, and seminal plasma immune responses [17]. EAV also infects monocytes, macrophages, and a small subpopulation of CD3^+^ T lymphocytes in equine peripheral blood mononuclear cells (PBMCs) in vitro [18,19,20]. We have also demonstrated that CD3^+^ T cell infection is mediated by a specific allele (CXCL16^S^) of the equine C-X-C motif chemokine ligand 16 (EqCXCL16) protein that renders these cells susceptible to EAV infection in vitro [21]. In contrast, CD3^+^ T cells from horses homozygous for the resistant allele of the EqCXCL16 (CXCL16^r^) are resistant to EAV infection in vitro [22]. Furthermore, we have demonstrated that stable transfection of HEK-293 cells with equine CXCL16^S^ increases susceptibility to EAV infection, confirming that EqCXCL16S can act as a cell entry receptor for EAV [21]. Moreover, EAV also preferentially infects subpopulations of horse CD14+ monocytes expressing EqCXCL16S, and that infection of these cells is significantly reduced by pretreatment with anti-EqCXCL16 polyclonal antibodies raised in guinea pig [22]. Interestingly, a population of CD14^+^ cells derived from horses homozygous for the CXCL16^r^ allele is also susceptible to EAV infection, indicating that other cellular receptors are involved in virus entry. EqCXCL16 differs substantially from CXCL16 orthologues in other species [22], indicating that non-equine CXCL16 sequences are unlikely to function as entry receptors. However, it is well-documented that a wide variety of continuous cell lines from different mammalian species other than equids (which obviously do not express equine CXCL16) support EAV replication, including African green monkey kidney cells (Vero and MA-104), baby hamster kidney fibroblasts (BHK-21), normal rabbit kidney epithelial (RK-13) cells, HeLa cells, and mouse connective tissue cells (L-M) [9,23,24,25,26,27,28,29,30,31]. In addition, it has been demonstrated that heparin treatment partially inhibits EAV infection of RK-13 cells, suggesting that heparin may play a role in EAV binding; however, it is not the only critical factor [32,33]. Recently, CD81 from equine and other species was identified as another cellular receptor for EAV in vitro [31]. These data suggest the involvement of other receptors(s) or cofactors(s) as determinants of EAV cell tropism. Therefore, the goal of this study was to identify additional receptor(s) or accessory cell proteins involved in EAV infection in vitro.
2. Materials and Methods
2.1. Cells
Equine pulmonary artery endothelial cells (EECs) [34,35,36] were propagated in Dulbecco’s modified Eagle’s medium (DMEM with 4.5 g/L glucose, sodium pyruvate, and L-glutamine; Corning, New-York, NY, USA) supplemented with 10% ferritin-supplemented bovine calf serum (Hyclone Laboratories LLC, Logan, UT, USA), 1 mM sodium pyruvate (Gibco, Waltham, MA, USA), 2 mM L-glutamine (Gibco), and 0.1 mM non-essential amino acids (NEAA; Gibco). Equine dermal fibroblasts (E. Derm, NBL-6 ATCC^®^ CCL-57, American Type Culture Collection [ATCC^®^], Manassas, VA, USA) and the human breast cancer cell line MCF7 (ATCC^®^ HTB-22) were propagated in Eagle’s minimal essential medium (EMEM; with Earle’s salts and L-glutamine; Corning) supplemented with 10% fetal bovine serum (FBS), 1 mM of sodium pyruvate and 0.1 mM NEAA. Baby hamster kidney (BHK-21, C-13, ATCC^®^ CCL-10), and high-passage rabbit kidney cells (HP-RK-13 [KY] P399-409, originally derived from ATCC CCL-37^TM^) were propagated in EMEM supplemented with 10% ferritin-supplemented bovine calf serum. The human colorectal adenocarcinoma cell line DLD-1 (ATCC^®^ CCL-221) was propagated in RPMI 1640 (Gibco) supplemented with 10% FBS and 2 mM L-glutamine. Human embryonic kidney cells (HEK-293, ATCC^®^ CRL-1573) were propagated in DMEM supplemented with 10% FBS and 2 mM L-glutamine. All media contained 100 IU/mL penicillin, 100 µg/mL streptomycin (Gibco), and 0.25 µg/mL amphotericin B (Gibco). Cells were cultivated in a humidified incubator at 37 °C and 5% CO_2_.
2.2. Viruses
Two strains of EAV, the virulent Bucyrus strain (VBS; ATCC ^®^ VR-796) [3,4] and the recombinant EAV VBS strain expressing mCherry (EAV sVBSmCherry) [37], were used in this study. Both viruses were used at their second passage on EECs, and high-titer stocks were generated as previously described [22,35].
2.3. Antibodies and Chemical Compounds
Mouse monoclonal antibodies specific to EAV GP5, N and nsp1 (6D10, 3E2, and 12A4, respectively) have been described previously [38,39]. Goat anti-rabbit IgG (H + L)-HRP and goat anti-mouse IgG (H + L)-HRP were purchased from Cell Signaling Technology (Danvers, MA, USA) and Southern Biotech (Birmingham, AL, USA). Mouse anti-vimentin MAb (Clone V9) and chicken anti-vimentin polyclonal antibody (PA1-10003) were purchased from Invitrogen (Waltham, MA, USA). Normal chicken IgY isotype control (AB-101-C) was purchased from R&D Systems (Minneapolis, MN, USA). Goat anti-mouse IgG (H + L) conjugated to Alexa Fluor (AF) 488, Goat anti-mouse IgG (H + L) conjugated to AF 647 were purchased from Life Technologies (Grand Island, NY, USA). Withaferin A (HY-N2065; MedChemExpress, Monmouth Junction, NJ, USA) was dissolved in DMSO (Sigma-Aldrich, Saint-Louis, MO, USA) at 10 mM and stored at −20 °C until used.
2.4. Virus Overlay Protein Binding Assay and Far-Western Blot
One hundred micrograms of total protein lysate from EECs and E. Derm cells or 30 μg of membrane or cytoplasmic protein fractions from EECs were separated in 12% SDS-PAGE and transferred onto a polyvinylidene fluoride (PVDF; Bio-Rad, Hercules, CA, USA) membrane for Far-Western blot (Far-WB) analysis following a modification of the published protocol [21,22]. The bound proteins were then denatured and gradually renatured on the membrane by sequential incubation with 6 M, 3 M, 1 M, and 0.1 M guanidine-HCl in freshly prepared AC buffer (100 mM NaCl, 20 mM TRIS [pH 7.5], 10% glycerol, 0.5 mM Ethylenediaminetetraacetic Acid [EDTA], 0.1% Tween-20, 2% non-fat dry milk, and 5 mM DTT) for 30 min at room temperature or with only AC buffer in the absence of guanidine-HCl overnight at 4 °C. The membrane was blocked with 5% non-fat dry milk in TBS-T (0.1% Tween-20) and overlaid with purified wild-type EAV VBS (15 μg/mL) and incubated overnight at 4 °C. The next day, the membrane was washed vigorously three times for 10 min and incubated with the mouse MAb (6D10) directed against EAV GP5 envelope glycoprotein. MAb binding was detected using a goat anti-mouse IgG conjugated to HRP and the ECL-detection system using SuperSignal^®^ West Pico chemiluminescent substrate (Thermo Fisher Scientific, Waltham, MA, USA).
2.5. Separation of Cytoplasmic and Membrane Protein Fractions
EECs were washed in ice-cold 1× phosphate-buffered saline (PBS), scraped, and then pelleted at 300 × g for 5 min. Cytoplasmic and membrane protein extracts were prepared using the Mem-PER^TM^ Plus Membrane Protein Extraction Kit (Thermo Fisher Scientific) following the manufacturer’s protocol. Briefly, the cell pellet was washed twice with Cell Wash solution and resuspended in permeabilization buffer, vortexed to get a homogeneous cell suspension. After incubating at 4 °C for 10 min, cells were centrifuged at 16,000× g for 15 min to collect the supernatants containing cytosolic proteins. The pellet was resuspended in solubilization buffer and incubated at 4 °C for 30 min with constant mixing, then pelleted again at 16,000 × g for 15 min at 4 °C. The supernatants containing cytosolic proteins and the membrane and membrane-associated proteins were stored at −80 °C until used.
2.6. Protein Trypsin Digestion and LC-MS/MS Analysis
EECs were solubilized, and integral membrane proteins and membrane-associated proteins were enriched and separated from cytoplasmic proteins using the Mem-PER^TM^ Plus Membrane Protein Extraction Kit (Thermo Fisher Scientific) as described above. One hundred micrograms of membrane and cytoplasmic proteins were subjected to 12% SDS-PAGE separation. The protein gel band at ~57 kDa was excised and subjected to dithiothreitol reduction, iodoacetamide alkylation, and in-gel trypsin digestion using a standard protocol, and the tryptic peptides were subjected to shotgun proteomics analysis as previously described [40]. The resulting tryptic peptides were extracted, concentrated to 15 µL using a SpeedVac, and 3 µL was injected for nano-liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis using an LTQ-Orbitrap mass spectrometer (Thermo Fisher Scientific) coupled with an Eksigent Nanoflex cHiPLC™ system (Eksigent, Dublin, CA, USA) through a nano-electrospray ionization source. The peptide samples were separated with a reversed phase cHiPLC column (75 μm × 150 mm) at a flow rate of 300 nL/min. Mobile phase A was water with 0.1% (v/v) formic acid, while B was acetonitrile with 0.1% (v/v) formic acid. A 50 min gradient condition was applied: initial 3% mobile phase B was increased linearly to 50% in 24 min and further to 85% and 95% for 5 min each before it was decreased to 3% and re-equilibrated. The mass analysis method consisted of one segment with eight scan events. The first scan event was an Orbitrap MS scan (100–1600 m/z) with 60,000 resolutions for parent ions, followed by data-dependent MS/MS for fragmentation of the seven most intense ions with the collision-induced dissociation (CID) method. The LC-MS/MS data were submitted to a local mascot server for MS/MS protein identification via Proteome Discoverer^TM^ (version 1.3, Thermo Fisher Scientific) against Equus caballus of the NCBI database (NCBI: txid9796). Parameters used in the MASCOT MS/MS ion search were trypsin digest with a maximum of two miscleavages, cysteine carbamidomethylation, methionine oxidation, a maximum of 10 ppm MS error tolerance, and a maximum of 0.8 Da MS/MS error tolerance. A decoy database was built and searched. Filter settings that determine false discovery rates (FDR) are used to distribute the confidence indicators for the peptide matches. Peptide matches that pass the filter associated with the strict FDR (with a target setting of 0.01) are assigned as high confidence. For MS/MS ion search, proteins with two or more high-confidence peptides were considered unambiguous identifications without manual inspection. Proteins identified with one high-confidence peptide were manually inspected and confirmed.
2.7. Time-Course Infection
Approximately 3 × 10^5^ cells (EECs, E. Derm, HP-RK-13, BHK-21, HEK-293, DLD-1, and MCF7) were seeded in 24-well plates and incubated at 37 °C and 5% CO_2_ in a humidified incubator. After 24 h, the supernatant was removed, and cells were infected with EAV sVBSmCherry at a multiplicity of infection (MOI) of 1 for 1 h at 37 °C. After three washing steps with 1× PBS, 500 μL of medium was added, and the plates were returned to the incubator. The plates were frozen and thawed three times at 12, 24, and 48 h post-infection (hpi), and the cell lysate was collected for virus titration. A total of 3 × 10^5^ HEK-293, HEK_CTL, and HEK_EqVIM seeded in 24-well plates and infected with EAV sVBSmCherry were placed in an IncuCyte^®^ Live Cell Analysis System (Sartorius, Göttingen, Germany), and the phase area and red area were recorded every six hours. The percentage of red area divided by the phase area was calculated using the IncuCyte^®^ 2022B Rev2 software.
2.8. Virus Titer Determination by Plaque Assay
Cell lysates were ten-fold diluted and inoculated on HP-RK-13 cells in a 12-well plate format. After 1 h of adsorption at 37 °C, cells were overlaid with complete EMEM containing 0.75% carboxymethylcellulose (Sigma-Aldrich) and incubated for 96 h at 37 °C and 5% CO_2_ in a humidified incubator. Media was removed, cells were subsequently fixed and stained with 0.2% crystal violet solution (Sigma-Aldrich) in 10% neutral buffered formalin for 3 h, and subsequently rinsed with tap water. Plaques were counted, and viral titers were determined as PFU per ml.
2.9. Blocking Cell Surface Vimentin with Anti-Vimentin Polyclonal Antibody and Withaferin A
Approximately 3 × 10^5^ of EECs were seeded in 24-well plates and incubated at 37 °C and 5% CO_2_ in a humidified incubator. After 24 h, the supernatant was removed, and cells were pre-treated with 10, 20, 40, and 80 µg/mL of vimentin polyclonal antibody (PA1-10003) or with equivalent concentrations of an isotype control for 1 h at 37 °C, diluted in fresh medium without FBS. Another set of cells was also pre-treated with 0.25, 0.5, 1, and 2 µM of Withaferin A (WFA) or DMSO (Sigma-Aldrich) diluted in fresh medium without FBS for 4 h at 37 °C. After three washing steps with 1× PBS, the cells were infected with EAV sVBSmCherry at an MOI of 0.1 for 1 h at 37 °C. After three washing steps with 1× PBS, fresh complete medium was added, and cells were incubated at 37 °C for 24 h. The cytotoxicity effect of WFA (0.0625 µM to 32 µM) on EECs was determined using CellTiter-Glo^®^ (Promega, Madison, WI, USA) at 24 h treatment as previously described [41].
2.10. Establishment of Stable HEK Cells Expressing Equine Vimentin
A stable cell line expressing equine vimentin (EqVim) was generated as described previously [21], with minor modifications. Briefly, for expression of EqVim protein in HEK-293 cells, a codon-optimized full-length EqVim sequence (NCBI Reference Sequence: NM_001243145.1) was commercially synthesized and cloned into the pIRESpuro3 plasmid (Takara Bio USA, San Jose, CA, USA) and named pIRESpuro3_EqVIM. In this construct, both EqVIM and puromycin-resistance sequences are expressed under the human cytomegalovirus immediate-early promoter and are separated by an internal ribosome entry site (IRES), enabling the co-expression of the two proteins from a single messenger RNA. An empty pIRESpuro3 vector, containing only the puromycin-resistance sequences, was used as a negative control (pIRESpuro3_CTL). Plasmid stocks were prepared by the transformation of original plasmids into chemically competent DH5α E. coli (Thermo Fisher Scientific) by the standard heat shock protocol. Bacteria were grown overnight on sterile LB broth supplemented with 100 mg/mL ampicillin. Plasmids were purified using the Plasmid Plus Maxiprep Kit (Qiagen, Valencia, CA, USA). HEK-293 cells seeded in 6-well plates (2 × 10^6^ cells/well) were transfected with 3 μg of pIRESpuro3_EqVIM and pIRESpuro3_CTL using Lipofectamine 3000 (Life Technologies, Grand Island, NY, USA), in serum-free conditions, according to the manufacturer’s instructions. At 48 h post-transfection, the medium was replaced with fresh medium containing 4 µg/mL of puromycin (Gibco). Culture media was replaced every other day until only the puromycin-resistant colonies were selected. These puromycin-resistant cells were cloned by limiting dilution in 96-well plates and screened by IFA to identify clones expressing a high level of EqVim protein expression. All the experiments in HEK_EqVim cells were performed within passages 7 to 10.
2.11. Indirect Immunofluorescence Assay (IFA)
EECs, E. Derm, HEK293, DLD-1, and MCF7 were seeded in 24-well plates containing cover slips (VWR, Radnor, PA, USA). Cells were washed in cold 1× PBS (pH 7.4) and fixed in 4% paraformaldehyde (PFA) (Sigma Aldrich, St. Louis, Missouri, USA) for 30 min at room temperature (RT). Cells were then stained following our previously published protocol [42]. Briefly, the cells were then washed three times with cold 1× PBS containing 10 mM glycine (PBS-Glycine; Sigma Aldrich) and permeabilized with 0.2% Triton X-100 (Sigma Aldrich) diluted in 1× PBS for 10 min or without detergent when examination of surface staining was required. Cells were washed in PBS-glycine and blocked with 5% FBS for 30 min at RT before incubation with specific primary antibodies (vimentin MAb, Clone V9; 1:400 dilution; mouse-anti EAV nsp1, Clone 12A4, 1:800 dilution) for 1 h at 37 °C. Cells were washed and incubated with conjugated secondary antibodies for 40 min at RT. Hoechst 33,342 (Invitrogen) solution was added to each well for an additional 20 min for nuclear staining (final concentration: 2 µg/mL). After washing, the coverslip was mounted on a glass slide using Fluoromount-G^TM^ mounting medium (SouthernBiotech, Birmingham, AL, USA). Fluorescence was examined using an Olympus FV300 confocal microscope (Hachioji, Tokyo).
2.12. SDS PAGE and Western Immunoblotting
HP-RK13, EECs, E. Derm, BHK-21, HEK-293, DLD-1, and MCF7 were lysed in 1× RIPA lysis buffer (Santa Cruz Biotechnology, Dallas, TX, USA) containing Halt phosphatase inhibitor cocktails (Thermo Fisher Scientific) and 1 mM of Phenylmethanesulfonyl Fluoride (PMSF). After 10 min of incubation on ice, the protein lysates were collected and centrifuged at 14,000× g for 10 min at 4 °C, and the supernatant was collected and stored at −80 °C until used. The solubilized proteins were mixed with Pierce 5× Lane reducing marker sample buffer containing 100 mM dithiothreitol (DTT) (Thermo Fisher Scientific). Samples were resolved in SDS-polyacrylamide gel (5% stacking and 12% resolving) at 200 V for 30 to 45 min and then transferred onto a PVDF membrane at 25 V, 2.5 A for 7 min using the Trans-Blot Turbo Transfer System (Bio-Rad, Hercules, CA, USA). The membrane was blocked with EveryBlot Blocking Buffer (Bio-Rad) for 10 min at RT and incubated with primary antibodies (Abs), (mouse anti-vimentin MAb (Clone V9) [1:1000], rabbit anti-β-actin [1:1000], and mouse anti-EAV GP5 MAb [6D10, 1:2000] diluted in EveryBlot Blocking Buffer overnight at 4 °C. The following day, the membranes were washed with TBS-T (Bio-Rad) and then incubated with secondary antibodies (anti-rabbit, or anti-mouse IgG, as appropriate), conjugated with horseradish peroxidase (HRP, 1:3000) (Cell Signaling, Danvers, MA, USA), diluted in EveryBlot Blocking Buffer for 1 h at RT. The membranes were washed again, and antibody binding was visualized with a ChemiDoc Imaging System (Bio-Rad) using Clarity Western ECL Substrate (Bio-Rad). Relative protein expression was quantified by densitometric analysis of Western blot bands using ImageJ (version 1.54d) software. The intensity of each target protein band was normalized to that of the corresponding loading control (β-actin) to correct for variations in protein loading. The normalized values were then expressed relative to the control sample, which was set to 1, to determine the fold change in protein expression across samples.
2.13. Flow Cytometry Analysis
HP-RK13, EECs, E. Derm, BHK-21, HEK-293, DLD-1, and MCF7 were harvested and washed with Fluorescence-activated cell sorting (FACS) buffer (1× PBS containing 0.1% sodium azide (VWR) and 1% heat-inactivated sterile-filtered goat serum (EquiTech-Bio, Inc., Kerrville, TX, USA)). Subsequently, cells were fixed with 4% PFA and labeled for 30 min at 4 °C with mouse anti-vimentin MAb (Clone V9; 1:400 dilution in FACS buffer), washed three times with FACS buffer, and stained with goat anti-mouse IgG (H + L) conjugated to AF 488 (1:400 dilution in FACS buffer). After three additional washing steps, flow cytometry data were collected using a BD FACS Calibur™ cytometer with 10^5^ events and analyzed with FlowJo version 10 software (BD Biosciences).
2.14. Equine CXCL16 Genotyping Determination
A total of 5.0 × 10^6^ E. Derm and EECs were collected and subjected to DNA extraction using DNeasy Blood & Tissue Mini Kit (Qiagen, Valencia, CA, USA), following the manufacturer’s instructions. Equine CXCL16 genotype was determined using the custom TaqMan^®^ SNP Genotyping Assay developed by our laboratory [43].
2.15. Statistical Analysis
Statistical analyses were performed using GraphPad Prism v10.2.3 statistical analysis software (GraphPad, San Diego, CA, USA). Data with a normal distribution were analyzed using one-way or two-way ANOVA followed by Dunnett’s post hoc test, while non-normally distributed data were evaluated using the Friedman non-parametric test with Dunn’s post hoc test. Data are expressed as mean ± standard deviation (SD). * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001.
3. Results
3.1. A 57 kDa Host Cell Membrane Protein Specifically Interacts with EAV
Our previous studies showed that CXCL16S protein (CXCL16^S/S^, CXCL16^S/r^) functions as an entry receptor for EAV [21,22]. Thus, two cell lines, EECs and E. Derm cells, susceptible to EAV infection, were first genotyped for EqCXCL16. Results show that EECs are heterozygote for CXCL16 (CXCL16^S^^/^^r^), and E. Derm cells are homozygote for the resistant allele (CXCL16^r/r^). The susceptibility of E. Derm cells with CXCL16^r/r^ genotype led us to hypothesize that EAV uses different molecule(s) as entry receptors to infect E. Derm cells in vitro. To identify the EAV interacting proteins, a Far-Western blot (Far-WB) in combination with virus overlay protein binding assay (VOPBA) was performed using both EECs and E. Derm cells. Cell lysates from EECs and E. Derm cells were separated by electrophoresis through an SDS-polyacrylamide gel and transferred to a solid matrix support (PVDF membrane) before incubation with purified live EAV. The positions of the EAV binding proteins were determined after the incubation of the membrane with an anti-EAV GP5 MAb (anti-GP5, 6D10 [39]) and an HRP-conjugated anti-mouse IgG secondary antibody (Figure 1A). The results showed that both EECs and E. Derm cell lysates had a common protein with an approximate molecular weight of 57 kDa that specifically interacts with EAV. On the other hand, when the membrane was incubated with the same antibodies in the absence of purified EAV, no interacting protein was observed, confirming the specificity of the protein interacting with EAV but not with other cellular proteins (Figure 1B).
To further identify the cellular localization of this EAV binding protein, sub-cellular fractionation of EECs was performed to separate membrane and cellular proteins, and 30 µg of each fraction was separated by SDS-PAGE and transferred onto a PVDF membrane, and Far-WB coupled to VOPBA was performed. The results showed that the same 57 kDa protein that binds to EAV was present on the membrane, but not the cytoplasmic fraction of EECs (Figure 1C). When a similar membrane was incubated with the same antibodies, but in absence of purified EAV, no band was observed, confirming the specificity of the protein interacting with EAV but not with other cellular proteins (Figure 1D). Interestingly, no binding protein with a molecular weight of 30 kDa (unglycosylated CXCL16), or 35–40 kDa (glycosylated CXCL16) was observed to bind EAV. Altogether, these results show that EAV binds to a 57 kDa protein located at the membrane of the EECs and E. Derm cells.
3.2. Identification of Vimentin as EAV-Interacting Host Cell Protein
Next, to investigate EAV-interacting proteins, cytoplasmic and membrane protein fractions from EECs were separated using two denaturing polyacrylamide gels. One gel was stained with Coomassie Brilliant Blue to visualize protein bands (Figure 2A), while the other was used for Virus Overlay Protein Binding Assay (VOPBA) and Far-Western blotting to identify potential EAV-binding proteins. A distinct band of approximately 57 kDa was detected in the membrane protein fraction (1A; Figure 2A), whereas no such band was observed in the cytoplasmic fraction (2A; Figure 2A). To identify this protein, the Coomassie-stained gel of EEC membrane proteins was aligned with the VOPBA membrane, and the band corresponding to the 57 kDa region was carefully excised, processed, and analyzed by LC-MS/MS. The identity of the protein was then determined by using the homology search tool, MASCOT, where the mass/charge values were matched with the information available in the database [44]. A representative MS/MS of a selected peptide LGDLYEEEMR was shown (Figure 2B). The analysis revealed that the purified 57 kDa EAV-interacting protein showed high similarity to vimentin, with a MASCOT score of 8398.8 and a peptide sequence coverage of 89.27% (Figure 2C–E). EECs and E. Derm cells were then stained for cell surface and intracellular vimentin using an anti-vimentin MAb (Clone V9). Results show that both cell lines express not only intracellular vimentin but also cell surface vimentin at the membrane (Figure 2F,G). Altogether, these results suggest that EAV interacts specifically with the vimentin protein.
3.3. Vimentin Expression Is a Determinant of EAV Susceptibility
To determine whether vimentin plays a critical role in EAV infection in vitro, cell lines derived from both horse (EECs, E. Derm) and other mammalian species (BHK-21, HP-RK-13, HEK-293, MCF7, and DLD-1) were infected with EAV sVBSmCherry at an MOI of 1, and viral replication was assessed at 12, 24, and 48 hpi through freeze–thaw lysis and subsequent virus titration. Results indicated that EECs, E. Derm, BHK-21, and RK-13 were highly susceptible to EAV infection and supported virus replication (Figure 3A). HEK-293 exhibited intermediate susceptibility with lower viral titers (≈3 Log_10_ difference at 24 hpi and ≈1.5 Log_10_ difference at 48 hpi, when compared to EECs), suggesting that HEK-293 cells restrict multiple rounds of viral replication. MCF7 and DLD-1 did not support EAV replication. Infected EECs, E. Derm, DLD-1, and MCF7 were stained for IFA at 12 hpi, allowing EAV to perform one cycle of replication (Figure 3B). High levels of nsp1 protein and mCherry were detected in both EECs and E. Derm. In contrast, neither nsp1 nor mCherry expression was observed in DLD-1 or MCF7 cells, confirming that these cell lines do not support EAV replication. Total vimentin expression was then assessed by Western blotting (Figure 3C,D), and cell surface vimentin expression was evaluated by flow cytometry (Figure 3E). High levels of total and cell surface vimentin were observed in HP-RK13, EECs, E. Derm, and BHK-21 cells, while HEK-293 cells expressed lower levels. DLD-1 and MCF7 cells showed no detectable vimentin expression (p = 0.008 and p = 0.015 for DLD-1 and MCF7 when compared to EECs, respectively) (Figure 3D). The absence of vimentin expression in DLD-1 and MCF7 was also confirmed by IFA (Figure 3F). To evaluate if vimentin expression is required for EAV to bind the target cells, a virus-binding assay was performed. EECs, E. Derm, DLD-1, and MCF7 were infected with EAV sVBSmCherry at an MOI of 1. After 1 h of adsorption at 4 °C, the cells were rinsed five times to remove unbound particles and subsequently frozen. After three cycles of freezing and thawing, bound infectious particles were titrated. The results show that EAV binds at similar levels to EECs, E. Derm, DLD-1, and MCF7 (Figure 3G), indicating that vimentin is not involved in the initial binding process but instead acts during a later stage of infection. Finally, alignment of vimentin protein sequences from susceptible equine species (Equus caballus and Equus asinus) and from susceptible cell line species (Homo sapiens, Chlorocebus aethiops, Oryctolagus cuniculus, Mus musculus, and Mesocricetus auratus) revealed a high degree of conservation across mammals, with sequence identity ≥96.8% (Figure 3H). These findings suggest that vimentin expression correlates with cellular susceptibility to EAV infection.
3.4. Pre-Treatment of EECs with Anti-Vimentin Polyclonal Antibody and Withaferin Inhibited EAV Replication
EECs were pre-treated with anti-vimentin polyclonal antibodies at different concentrations (10, 20, 40, and 80 μg/mL) or with an isotype control (80 μg/mL). Subsequently, cells were washed and infected with EAV at an MOI of 0.1 for 1 h at 37 °C. At 24 hpi, cells were lysed by cycles of freezing/thawing, and virus titration was performed (Figure 4A). Results showed that all the tested concentrations of anti-vimentin polyclonal antibody reduced viral titers when compared to isotype-treated cells, and a significant reduction was observed for 20 µg/mL (p = 0.029) and 80 µg/mL (p = 0.022). On the other hand, a pre-treatment with an isotype control did not show any effect (p > 0.05). Cells were also examined by IFA at 12 hpi (Figure 4B). High levels of nsp1 protein and mCherry were detected in both untreated EECs and those treated with 80 µg/mL of the isotype control antibody. In contrast, treatment with 80 µg/mL of the anti-vimentin polyclonal antibody resulted in only a few cells staining positive for nsp1 and mCherry. These data indicate that blocking cell surface vimentin with a polyclonal anti-vimentin antibody reduces infectious titers, supporting a role for cell surface vimentin in facilitating EAV entry in vitro.
Withaferin-A (WFA), a steroidal lactone isolated from the plant Withania somnifera, has been shown to bind and alter the distribution of the vimentin intermediate filament in human and bovine endothelial cells in vitro [45]. Here, we evaluated the effect of WFA on vimentin conformation in EECs and its pre-treatment on EAV infection. First, a viability experiment demonstrated that WFA is highly toxic at 24 h post-treatment, starting from 8 µM and has a half maximum toxic concentration (IC_50_) of 6.31 ± 1.25 µM (Figure 5A). Immunofluorescent staining of EECs treated with WFA for 6 h demonstrated that 2 µM WFA disrupts intracellular vimentin organization, leading to its aggregation (Figure 5B) and a significant reduction in cell surface vimentin expression (p < 0.001; Figure 5C). At 1 µM, no reduction in cell surface vimentin expression and no aggregation of intracellular vimentin was observed (Figure 5B,C). EECs were then incubated for 6 h with different non-toxic concentrations of WFA (0.25, 0.5, 1, and 2 µM) and subsequently infected with EAV sVBSmCherry strain at an MOI of 0.1. At 24 hpi, mCherry expression was monitored using a live cell imaging microscope before cell lysis for virus titration. Results showed that 1 and 2 µM of WFA significantly reduce mCherry expression (p < 0.001; Figure 5D,E). Additionally, infectious titers in the cell lysate are significantly reduced following pre-treatment with 1 µM (p = 0.016) and 2 µM (p < 0.001) of WFA (Figure 5F). However, lower concentrations (0.25 µM and 0.5 µM) did not affect mCherry expression or viral titers (p > 0.05; Figure 5E,F). These results suggest that disruption of the vimentin’s conformation decreases the susceptibility of EECs to EAV infection.
3.5. Overexpression of Equine Vimentin in HEK-293 Cells Increases Cell Susceptibility to EAV
To further evaluate the role of the equine vimentin protein in EAV infection, we established a stable cell line expressing the equine orthologue of vimentin (EqVim) on naïve HEK-293 cells. HEK-293 cells were transfected with either pIRESpuro3_EqVIM or with the empty control vector pIRESpuro3_CTL and cultured in the presence of 4 µg/mL puromycin. A significantly higher expression of vimentin was observed by Western blotting in the HEK_EqVIM cells, when compared to the HEK-293 (p = 0.05), while no difference was observed for the HEK_CTL (Figure 6A). Similarly, an increase in intracellular and cell surface vimentin expression was observed by IFA on the HEK_EqVIM cells, when compared to the HEK-293 and HEK_CTL cells (Figure 6B). HEK-293, HEK_CTL, and HEK_EqVIM cells were then infected at an MOI of 1 with EAV sVBSmCherry and incubated at 37 °C in a live cell imaging microscope. Phase and red fluorescence (mCherry expression) were recorded every 6 h for 72 h and the ratio of mCherry area (%) to the phase area (%) was graphically represented in Figure 6C. Starting from 30 hpi, this ratio was significantly higher in HEK_EqVIM infected cells when compared to HEK-293 infected cells (p < 0.001), whereas no significant difference was detected in HEK_CTL cells (p > 0.05) for all time points. This disparity is visually apparent at 72 hpi, as illustrated in Figure 6D. The cell culture supernatant was also collected at 12, 24, 48, and 72 hpi and used for virus titration (Figure 6E). A significant increase in infectious particles was observed at 48 hpi (p = 0.034) and 72 hpi (p = 0.008) in the cell culture supernatant of HEK_EqVIM when compared to HEK-293, while no significant difference was observed for the HEK_CTL (p > 0.05). Concordant with the previous finding, this result shows that the presence of equine vimentin increases the susceptibility to EAV infection, confirming its important role in EAV infection in vitro.
4. Discussion
In previous studies, we have shown that a susceptible allelic variant of equine transmembrane CXCL16 (CXCL16S [EqCXCL16^S/S^ or EqCXCl16^S/r^]) acts as an entry receptor for EAV by binding with the virus and facilitating its internalization inside the host cells [21,22]. This susceptibility phenotype is associated with four non-synonymous mutations located in exon 1 of the EqCXCL16 gene. In contrast, the resistant variant, CXCL16R, found in horses lacking these mutations (EqCXCL16*^r/r^*), fails to bind EAV in vitro [22]. However, cells such as CD14+ monocytes from EqCXCL16^r/r^ horses and E. Derm, expressing only EqCXCL16R, are also susceptible to EAV infection. This led us to hypothesize that other host cell factors are involved in EAV infection. Recent work conducted by others identified CD81 and neonatal Fc receptor (FcRn) as EAV receptors [31,46].
Here, we demonstrated that vimentin, a type III intermediate filament protein [47,48], plays a determinant role in EAV infection. Vimentin has a molecular weight of 57 kDa and is expressed in various cells of mesenchymal origin, such as T lymphocytes, monocytes, fibroblasts, and endothelial cells [49]. Besides its structural role, vimentin may also be expressed at the cell surface [50,51]. Cell surface vimentin was reported as an attachment factor or co-receptor for various viruses belonging to the Nidovirales order, such as the related Porcine Reproductive and Respiratory Virus (PRRSV) [52], SARS-CoV [53] and SARS-CoV-2 [54,55], but also for viruses belonging to more distant orders, such as Zika virus [56], Dengue virus [57], Human Papillomavirus [58], Japanese Encephalitis virus [59,60], and Enterovirus 71 [61]. In this study, cell surface equine vimentin was identified to play a role in the attachment and infection of EAV. We first identified a 57 kDa protein expressed in the cell membrane of EECs as a receptor for EAV, which was further identified as vimentin by LC-MS/MS analysis. Consistent with our previous findings showing that EAV infects and persists in the stallion ampullae with a specific tropism for vimentin-positive stromal cells in the lamina propria, these results further support vimentin as a key host factor in EAV infection and persistence, potentially contributing to the maintenance of viral reservoirs in the reproductive tract [13]. Vimentin plays a multifaceted role in PRRSV infection, acting as both an attachment factor and an intracellular replication cofactor through interactions with the viral nucleocapsid and non-structural proteins [52,62,63]. Moreover, vimentin rearranges into cage-like structures around PRRSV replication complexes, supporting viral replication and suggesting a conserved role for vimentin in arterivirus persistence [64].
Surprisingly, no other EAV-binding proteins, such as EqCXCL16S, CD81, or FcRn, were detected by Far-Western blot in our assay, suggesting that vimentin may play a more prominent or direct role in viral attachment under these experimental conditions.
A clear correlation was observed between vimentin expression and cellular susceptibility to EAV infection. Although vimentin is highly conserved among mammalian species, EAV infection in vivo remains species-specific and restricted to equids, suggesting that its conserved nature primarily reflects a basic cellular role as an attachment factor rather than implying that EAV can utilize vimentin from other species as an entry receptor. In this study, we identified two cell lines, DLD-1 and MCF7, resistant to EAV infection. Notably, these cell lines do not express vimentin, as previously reported [65,66]. However, it is important to note that EAV can bind to DLD-1 and MCF7 at a similar level to EECs and E. Derm, demonstrating that vimentin is not involved in the initial binding process but instead acts during a later stage of infection (e.g., viral internalization or membrane fusion).
HEK-293 cells, which naturally express low levels of vimentin [67], supported only limited EAV replication. Overexpression of equine vimentin in these cells (HEK_EqVIM) significantly enhanced their susceptibility to infection, supporting the role of vimentin as a key attachment factor facilitating EAV entry. Overall, these findings support the role of vimentin as a key attachment factor facilitating EAV entry in vitro. While equine vimentin enhances susceptibility in heterologous cells, we have not directly shown that its expression alone is sufficient to confer permissiveness in vimentin-negative cells. Attempts to establish stable EqVIM expression in MCF7 and DLD-1 cells were unsuccessful. Alternative expression strategies, such as retroviral vector-mediated delivery, may be explored in future studies. Attempts to generate vimentin knockouts in EECs and E. Derm cells were unsuccessful due to cytotoxicity, highlighting the essential role of vimentin in maintaining cellular morphology. Alternative strategies, such as conditional knockdown, transient silencing, or the use of dominant-negative mutants, may help overcome this limitation and further clarify vimentin’s role in EAV infection.
Blocking the cell surface vimentin domain with vimentin polyclonal antibody decreases the early replication cycle of EAV. A similar observation was made when EECs were pre-treated with WFA, which caused vimentin conformational disruption, aggregation, and a reduction in cell surface vimentin expression. While the primary function of WFA is the alteration of vimentin conformation [45,68], it was also reported that WFA does not prevent the attachment and entry of Chikungunya virus, but acts as a viral protease (nsp2) inhibitor [69]. Therefore, while in line with the results observed with the vimentin polyclonal antibody, we cannot conclude that the decrease in viral replication and progeny virus production after WFA treatment is solely due to inhibition of cell surface vimentin expression. Further studies, such as assessing whether WFA interacts with EAV nsp2 or other viral proteins, will be required to clarify its mechanism of action.
Altogether, our findings indicate that cell surface equine vimentin plays a critical role in EAV infection in vitro and may function as an important attachment cofactor facilitating viral entry. Although the precise mechanism remains unclear, our data, together with prior studies on PRRSV and SARS-CoV-2 [52,54,70], suggest that vimentin may not act as a direct binding receptor but rather as a scaffold or accessory factor that stabilizes or enhances interactions between EAV and its primary receptors. Such receptors may include EqCXCL16S, CD81, FcRn, heparan sulfate, or other yet-unidentified surface molecules. Further studies, including co-immunoprecipitation and domain-mapping approaches, will be necessary to identify the vimentin domain(s) that interact with EAV, to determine whether vimentin directly engages EAV structural proteins, and to investigate its potential role in modulating the availability or function of the EAV receptor. In particular, analyses in E. derm cells could help clarify the interaction between vimentin and the yet-to-be-characterized EAV receptor.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Kuhn J.H. Lauck M. Bailey A.L. Shchetinin A.M. Vishnevskaya T.V. Bào Y. Ng T.F.F. Le Breton M. Schneider B.S. Gillis A. Reorganization and Expansion of the Nidoviral Family Arteriviridae Arch. Virol.201616175576810.1007/s 00705-015-2672-z 26608064 PMC 5573231 · doi ↗ · pubmed ↗
- 2Brinton M.A. Gulyaeva A.A. Balasuriya U.B.R. Dunowska M. Faaberg K.S. Goldberg T. Leung F.C.C. Nauwynck H.J. Snijder E.J. Stadejek T. ICTV Virus Taxonomy Profile: Arteriviridae 2021 J. Gen. Virol.202110200163210.1099/jgv.0.00163234356005 PMC 8513641 · doi ↗ · pubmed ↗
- 3Bryans J.T. Crowe M.E. Doll E.R. Mc Collum W.H. Isolation of a Filterable Agent Causing Arteritis of Horses and Abortion by Mares; Its Differentiation from the Equine Abortion (Influenza) Virus Cornell Vet.19574734113397177 · pubmed ↗
- 4Doll E.R. Knappenberger R.E. Bryans J.T. An Outbreak of Abortion Caused by the Equine Arteritis Virus Cornell Vet.195747697513397180 · pubmed ↗
- 5Balasuriya U.B.R. Carossino M. Timoney P.J. Equine Viral Arteritis: A Respiratory and Reproductive Disease of Significant Economic Importance to the Equine Industry Equine Vet. Educ.20183049751210.1111/eve.12672 · doi ↗
- 6Timoney P.J. Mc Collum W.H. Equine Viral Arteritis Vet. Clin. N. Am. Equine Pract.1993929530910.1016/S 0749-0739(17)30397-8PMC 71346768395325 · doi ↗ · pubmed ↗
- 7Balasuriya U.B.R. Snijder E.J. Heidner H.W. Zhang J. Zevenhoven-Dobbe J.C. Boone J.D. Mc Collum W.H. Timoney P.J. Mac Lachlan N.J. Development and Characterization of an Infectious c DNA Clone of the Virulent Bucyrus Strain of Equine Arteritis Virus J. Gen. Virol.20078891892410.1099/vir.0.82415-017325365 · doi ↗ · pubmed ↗
- 8Balasuriya U.B. Snijder E.J. van Dinten L.C. Heidner H.W. Wilson W.D. Hedges J.F. Hullinger P.J. Mac Lachlan N.J. Equine Arteritis Virus Derived from an Infectious c DNA Clone Is Attenuated and Genetically Stable in Infected Stallions Virology 199926020120810.1006/viro.1999.981710405372 · doi ↗ · pubmed ↗