Emerging Therapeutic Strategies in Prostate Cancer: Targeted Approaches Using PARP Inhibition, PSMA-Directed Therapy, and Androgen Receptor Blockade with Olaparib, Lutetium (177Lu)Vipivotide Tetraxetan, and Abiraterone
Piotr Kawczak, Tomasz Bączek

TL;DR
This review discusses new targeted treatments for advanced prostate cancer, including PARP inhibitors, PSMA-directed therapy, and androgen receptor blockers, and how they can be used effectively in clinical practice.
Contribution
The paper provides a clinically focused overview of integrating PARP inhibition, PSMA-directed RLT, and ARPIs in advanced prostate cancer treatment.
Findings
Olaparib benefits mCRPC patients with HRR mutations, especially BRCA1/2.
PSMA-directed RLT improves survival in PSMA-positive mCRPC after AR pathway inhibition.
Abiraterone remains a key therapy and can be combined with other agents for better outcomes.
Abstract
Prostate cancer is one of the most common malignancies in men, and advanced or metastatic disease remains associated with substantial morbidity and mortality. Therapeutic progress in recent years has been driven by the introduction of targeted treatment strategies, notably poly (ADP-ribose) polymerase (PARP) inhibitors, prostate-specific membrane antigen (PSMA)–directed radioligand therapy (RLT), and androgen receptor pathway inhibitors (ARPIs). This review summarizes evidence from phase II and III clinical trials, meta-analyses, and real-world studies evaluating the efficacy, safety, and clinical integration of olaparib, lutetium (177Lu) vipivotide tetraxetan, and abiraterone in advanced prostate cancer. Emphasis is placed on the practical clinical application of these agents, including patient selection, treatment sequencing, and combination strategies. PARP inhibition with olaparib…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
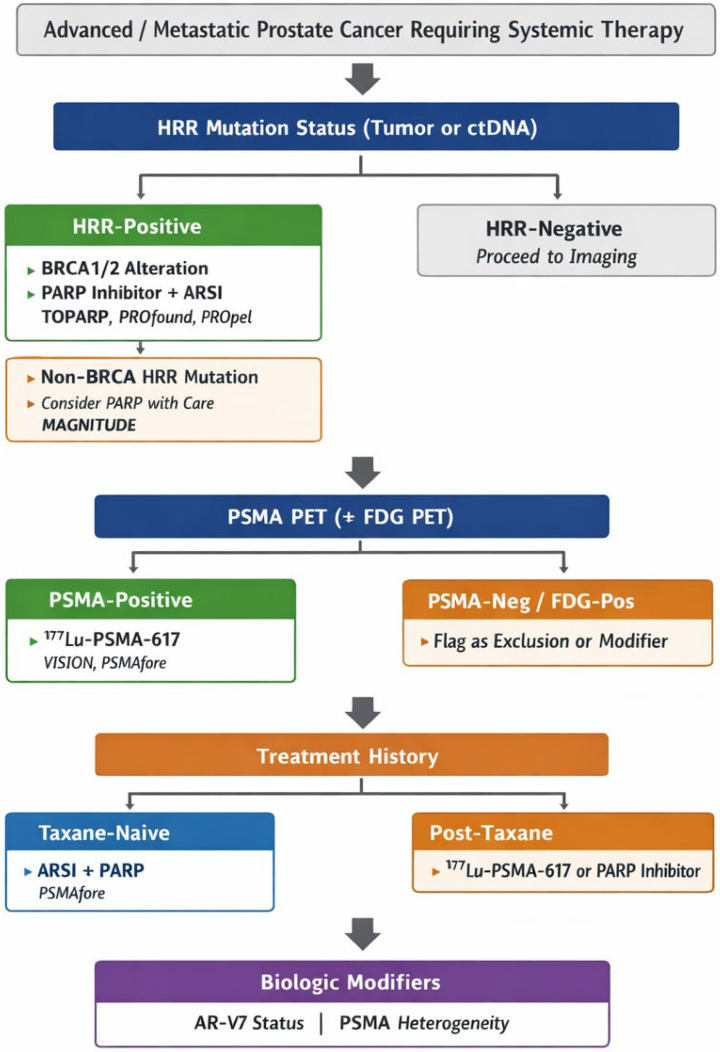 Figure 1
Figure 1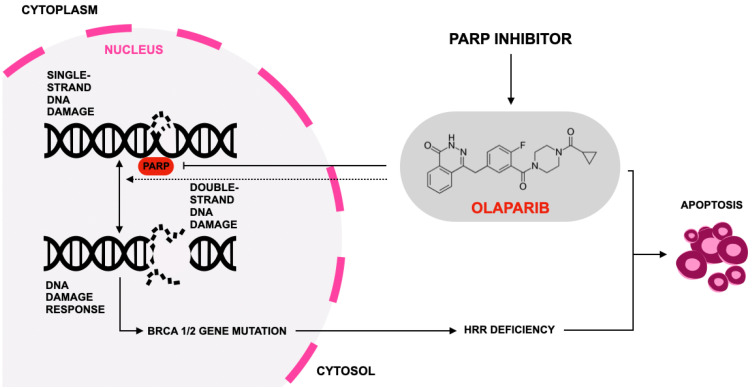 Figure 2
Figure 2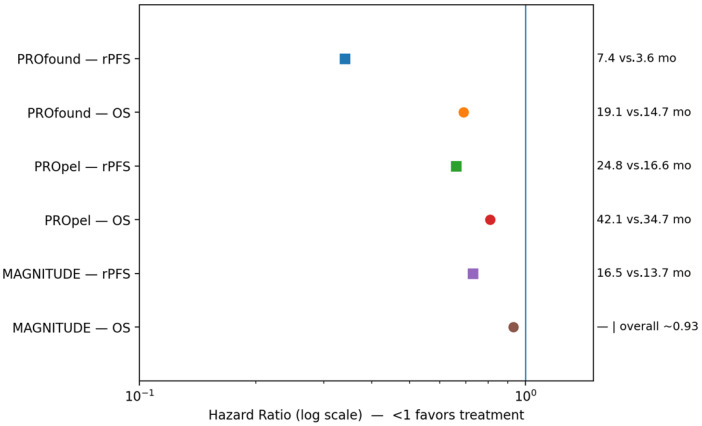 Figure 3
Figure 3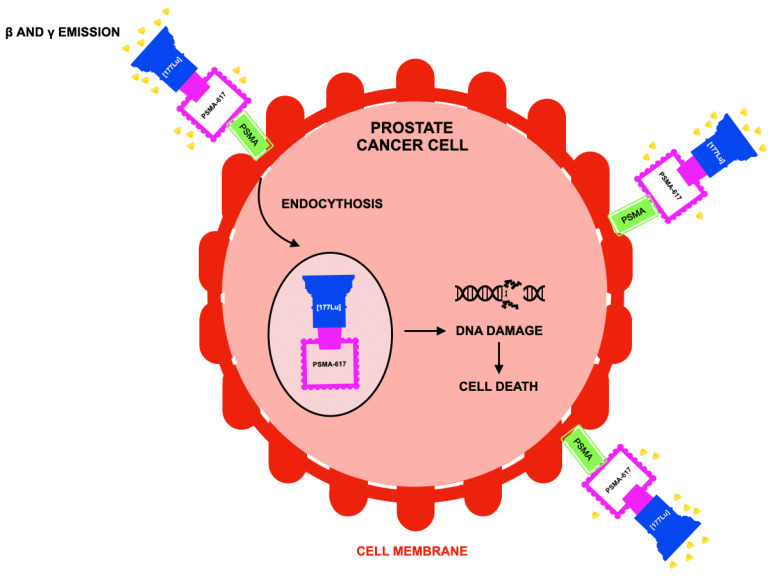 Figure 4
Figure 4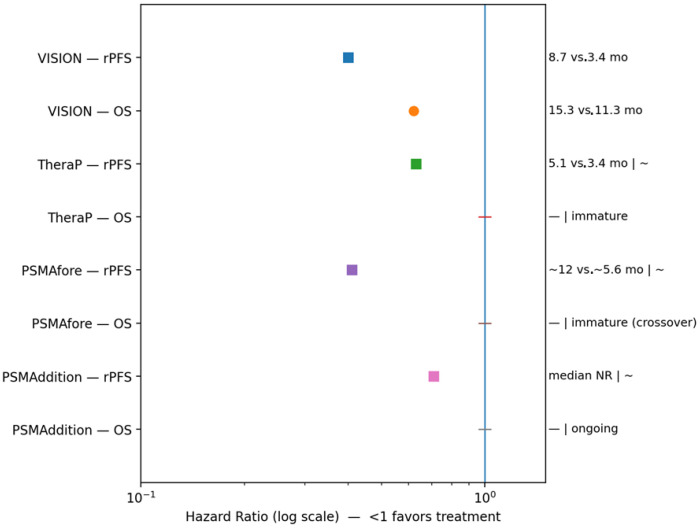 Figure 5
Figure 5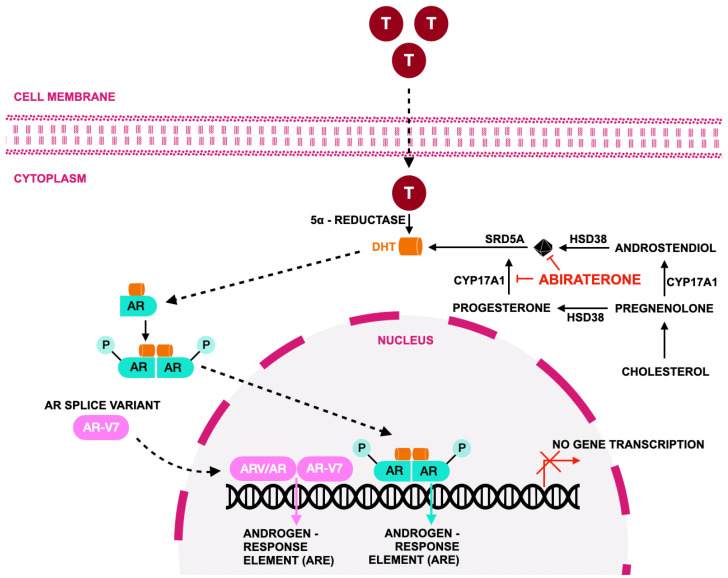 Figure 6
Figure 6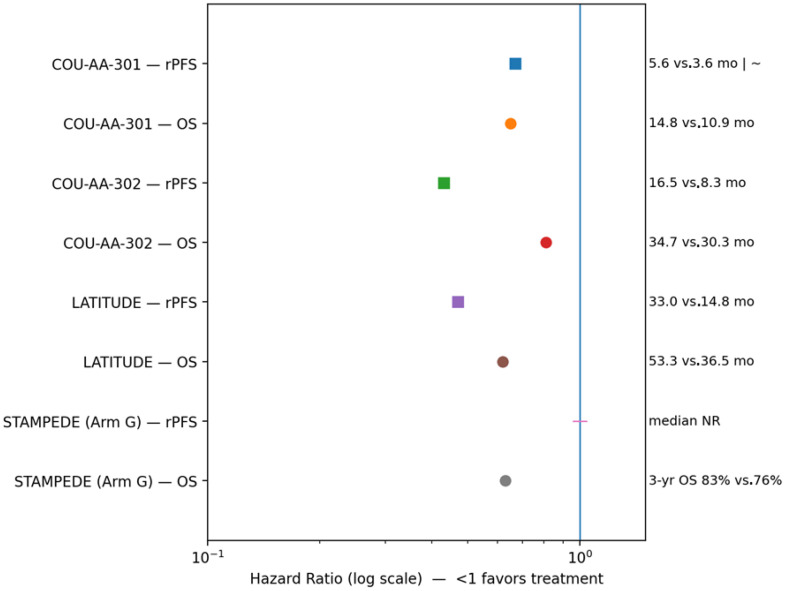 Figure 7
Figure 7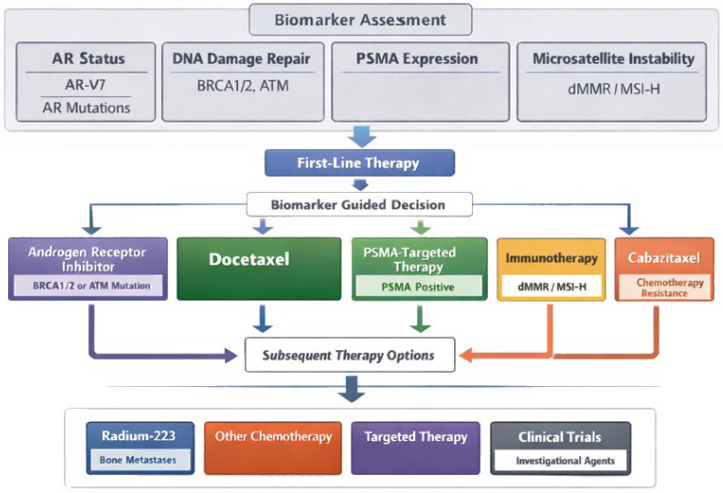 Figure 8
Figure 8Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsProstate Cancer Treatment and Research · PARP inhibition in cancer therapy · Prostate Cancer Diagnosis and Treatment
1. Introduction
Prostate cancer is the second most frequently diagnosed malignancy in men and remains a leading cause of cancer-related mortality worldwide [1,2,3]. While localized disease is often effectively managed with surgery or radiotherapy, advanced and metastatic prostate cancer continue to pose substantial therapeutic challenges. Disease progression typically follows a trajectory from hormone-sensitive prostate cancer to metastatic castration-resistant prostate cancer (mCRPC), a state characterized by persistent androgen receptor (AR) signaling despite castrate levels of testosterone. This transition is driven by a spectrum of molecular alterations, including AR amplification, intratumoral androgen synthesis, defects in DNA damage repair (DDR), and overexpression of prostate-specific membrane antigen (PSMA) [4,5,6,7].
Over the past decade, the therapeutic landscape of advanced prostate cancer has evolved rapidly, driven by the clinical translation of molecularly targeted strategies. Among these, three therapeutic pillars have emerged as central to contemporary management: (1) poly(ADP-ribose) polymerase (PARP) inhibition, particularly with olaparib, targeting tumors with homologous recombination repair (HRR) deficiencies; (2) PSMA-targeted radioligand therapy (RLT), most notably lutetium (^177^Lu) vipivotide tetraxetan (^177^Lu-PSMA-617), which enables targeted systemic radiation delivery; and (3) intensified AR pathway inhibition with agents such as abiraterone acetate, a CYP17A1 inhibitor that suppresses extragonadal androgen synthesis and enhances androgen deprivation therapy (ADT)–mediated AR blockade [8,9,10,11]. While these approaches are often discussed separately, their increasing convergence in clinical practice underscores the need for an integrative framework that considers biomarkers, disease biology, and treatment sequencing.
PARP inhibitors have fundamentally reshaped the treatment of HRR-deficient mCRPC. The pivotal PROfound trial demonstrated that olaparib significantly improved radiographic progression-free survival (rPFS) and overall survival (OS) in patients with BRCA1/2 and other HRR gene alterations following progression on AR signaling inhibitors (ARSIs) [12,13,14]. Subsequent analyses and real-world evidence reinforced the clinical importance of genomic profiling and supported regulatory approvals of olaparib for HRR-mutated mCRPC across multiple jurisdictions [15,16,17]. More recently, combination strategies such as olaparib plus abiraterone have been evaluated in first-line mCRPC. Trials including PROpel and MAGNITUDE have suggested potential activity beyond BRCA-mutated disease, while also highlighting important biological limitations and heterogeneity of benefit, particularly in biomarker-unselected populations [18,19,20].
SMA-targeted RLT represents another major advance, leveraging the high and relatively selective expression of PSMA in advanced prostate cancer. The phase III VISION trial established that ^177^Lu-PSMA-617, when added to standard of care, significantly improved OS and rPFS in patients with PSMA-positive mCRPC previously treated with ARSIs and taxane chemotherapy [21]. Earlier phase II data from the TheraP trial demonstrated superior prostate-specific antigen (PSA) response rates and a more favorable toxicity profile compared with cabazitaxel [22,23]. Subsequent real-world and registry-based studies have confirmed the reproducibility of these outcomes, supporting the incorporation of PSMA-RLT into clinical guidelines and routine practice [24,25].
AR pathway inhibition remains a cornerstone of therapy across the disease continuum. Abiraterone acetate, administered with prednisone, demonstrated significant OS benefits in both chemotherapy-naïve mCRPC (COU-AA-302) [26] and post-docetaxel mCRPC (COU-AA-301) [27]. Its role was further expanded by the LATITUDE and STAMPEDE trials, which established improved survival outcomes in high-risk metastatic castration-sensitive prostate cancer (mCSPC), thereby shifting treatment paradigms toward earlier and more intensive AR-axis suppression [28,29,30].
Despite these advances, critical clinical questions remain unresolved. Optimal sequencing of PARP inhibitors, PSMA-directed RLT, and next-generation AR pathway inhibitors has not been fully defined, particularly as these agents move into earlier lines of therapy. Combination strategies—including PARP inhibitors plus ARSIs or the integration of RLT with systemic treatments—are actively being explored, yet their appropriate application remains the subject of ongoing investigation and debate [31,32,33,34]. In parallel, real-world implementation is influenced by access to biomarker testing, PSMA PET imaging, and multidisciplinary expertise, raising important considerations regarding equity and generalizability of care [35,36]. Figure 1 illustrates the clinical decision-making algorithm for prostate cancer.
In this context, the present review aims to extend beyond existing narrative summaries by providing an integrative, biomarker-driven synthesis across these three therapeutic pillars, with a particular emphasis on therapeutic sequencing rather than isolated drug classes. We highlight how genomic profiling, functional imaging, prior treatment exposure, and toxicity considerations intersect to inform clinical decision-making. In addition, we discuss future directions and unresolved questions, including patient selection, combination strategies, and long-term safety. By framing these therapies within a unified clinical context, this review seeks to clarify their complementary roles and to support rational, personalized management of advanced prostate cancer.
This synthesis is based on a narrative literature review of PubMed and Scopus using the search terms “olaparib,” “lutetium (^177^Lu) vipivotide tetraxetan,” and “abiraterone” in combination with “targeted therapy” and “prostate cancer.” Peer-reviewed articles published between 2005 and 2025 were selected according to relevance, methodological rigor, and contribution to understanding therapeutic efficacy, mechanisms of action, resistance, and clinical integration. Both preclinical and clinical studies were included when mechanistic insights informed therapeutic strategy, enabling a comprehensive assessment of the benefits and limitations of targeted therapies in contemporary prostate oncology.
2. Olaparib
Olaparib is an orally available small-molecule inhibitor of PARP enzymes that has transformed treatment paradigms for tumors with defective HRR, most notably cancers harboring BRCA1/2 alterations; by binding the catalytic domain of PARP1 and PARP2, olaparib prevents PARylation at sites of single-strand DNA breaks, promotes trapping of PARP on DNA, converts replication-associated single-strand breaks into lethal double-strand breaks, and thereby induces synthetic lethality in cells deficient in HRR machinery (Figure 2) [37,38,39,40]. In clinical practice, this mechanistic specificity underpins the rationale for biomarker-guided use of olaparib and highlights the importance of accurate identification of HRR defects prior to treatment initiation.
The biochemical selectivity for PARP1/2 and the dual capacity of olaparib to inhibit catalytic activity and stabilize PARP–DNA complexes underlie both its antitumor efficacy and its class-typical toxicities, including myelosuppression and gastrointestinal adverse effects [42,43]. Early clinical development focused on BRCA-mutant ovarian and breast cancers, culminating in the first regulatory approvals in BRCA-mutated ovarian cancer in 2014 and establishing proof of principle for therapeutic exploitation of HRR deficiency [44,45]. Subsequent mechanistic and translational research broadened the understanding of PARP inhibition by elucidating additional modes of action—such as effects on replication-fork stability and immunomodulatory consequences—and by clarifying why specific HRR gene alterations (e.g., BRCA1/2) predict greater sensitivity than others (e.g., ATM), while also defining biologically plausible mechanisms of acquired resistance, including reversion mutations, restoration of end resection, replication-fork protection, and drug efflux [46,47,48]. These insights have direct implications for real-world treatment durability and for the strategic sequencing of therapies to delay or circumvent resistance.
Interest in olaparib for prostate cancer arose from the recognition that approximately 20–30% of mCRPC harbor deleterious germline or somatic alterations in HRR genes, defining a clinically actionable molecular subset [49,50]. Early clinical development of PARP inhibitors in this setting was characterized by exploratory, biomarker-unselected phase II studies, most notably TOPARP-A, which demonstrated that antitumor activity was strongly enriched among patients with DNA-repair defects. In TOPARP-A, objective responses and durable clinical benefit were concentrated in tumors harboring deleterious BRCA2, ATM, CHEK2, and PALB2 alterations, thereby validating a biomarker-led treatment strategy and establishing proof of concept for PARP inhibition in prostate cancer [42,51]. Parallel analyses confirmed meaningful improvements in rPFS and OS among HRR-altered tumors, directly informing the design of subsequent molecularly enriched trials [12,13].
Prospective validation followed in TOPARP-B, which restricted enrollment to patients with predefined HRR mutations and employed a randomized dose-comparison design, demonstrating higher response rates and greater clinical benefit in BRCA1/2-altered tumors compared with other HRR subgroups [14]. These findings were definitively confirmed in the phase III PROfound trial, which enrolled men with HRR-mutated mCRPC who had progressed on enzalutamide or abiraterone and used rPFS assessed by blinded independent central review as the primary endpoint. In PROfound, olaparib significantly improved rPFS compared with physician’s choice of androgen receptor–targeted therapy, increasing median rPFS from 3.0 to 7.4 months (absolute gain 4.4 months; HR 0.34; 95% CI: 0.25–0.47), and also prolonged OS (19.1 vs. 14.7 months; HR 0.69; 95% CI: 0.50–0.97), despite substantial crossover [12]. Benefit was particularly pronounced in patients with BRCA1/2 and ATM alterations, establishing PARP inhibition as a standard of care in this molecularly defined population and supporting regulatory approval in prostate cancer [42,44,52].
Regulatory milestones followed rapidly. On 19 May 2020, the U.S. Food and Drug Administration approved olaparib (LYNPARZA^®^) for adult patients with deleterious or suspected deleterious germline or somatic HRR gene–mutated mCRPC following progression on enzalutamide or abiraterone, with companion diagnostic assays specified to identify eligible patients; subsequent regulatory refinements expanded indications and enabled combination strategies in selected genomic subgroups [53,54,55]. These developments reinforced the practical necessity of integrating timely germline and somatic HRR testing into standard clinical workflows.
As clinical development progressed, attention increasingly shifted toward combination strategies and broader patient populations. Preclinical evidence of reciprocal interactions between AR signaling and DNA repair pathways provided a rationale for combining PARP inhibitors with androgen receptor signaling inhibitors (ARSIs). In this context, first-line randomized trials such as PROpel and MAGNITUDE evaluated PARP inhibitor–abiraterone combinations. In PROpel, olaparib plus abiraterone significantly prolonged rPFS compared with abiraterone alone (24.8 vs. 16.6 months; absolute gain 8.2 months; HR 0.66; 95% CI: 0.54–0.81), with final OS analysis showing a numerical improvement (42.1 vs. 34.7 months; HR 0.81; 95% CI: 0.67–1.00). Exploratory analyses suggested activity beyond BRCA1/2-mutated tumors; however, the magnitude of benefit was greatest in patients with HRR alterations, particularly BRCA mutations [26,56]. These findings supported FDA approval of the combination for BRCA-mutated mCRPC in 2023 [57,58,59].
In contrast, the MAGNITUDE trial prospectively stratified patients by HRR status and failed to demonstrate benefit in HRR-negative disease, leading to early closure of this cohort. This outcome underscored important biological and clinical distinctions between HRR-defined subpopulations and cautioned against extrapolating benefit to biomarker-unselected patients [18,19,20]. Collectively, these data indicate that while PARP inhibitor–ARSI combinations represent a significant therapeutic advance for patients with defined HRR alterations—particularly BRCA1/2 mutations—their routine use outside molecularly selected populations remains controversial [27,28].
Across clinical trials, efficacy has consistently correlated with specific HRR genotypes. Patients with BRCA1/2 alterations derive the most pronounced and durable improvements in rPFS and OS, whereas alterations in ATM, CHEK2, or CDK12 confer more variable and generally modest benefit. These genotype–phenotype associations have informed guideline recommendations advocating routine germline and somatic HRR testing in metastatic disease to guide PARP inhibitor use and familial risk counseling [47,60,61]. Implementation guidance has further addressed assay selection, interpretation of variants of uncertain significance, and concordance between tissue- and plasma-based testing modalities, all of which remain critical for real-world application [62].
The safety profile of olaparib is well characterized. Across pivotal trials including TOPARP-A, TOPARP-B, PROfound, and PROpel, grade ≥3 adverse events were reported in approximately 40–55% of patients, depending on line of therapy and combination partner [63,64,65,66]. Hematologic toxicity, particularly anemia, represented the most prominent severe adverse event, with grade ≥3 anemia occurring in approximately 20–25% of patients receiving olaparib monotherapy and 15–20% of those treated with olaparib plus abiraterone [67,68,69]. Fatigue and asthenia were common, although grade ≥3 events were generally infrequent (<10%), and gastrointestinal toxicities such as nausea and vomiting were typically manageable. These adverse events were often cumulative and frequently necessitated dose interruptions or reductions, underscoring the importance of routine hematologic monitoring and early supportive management, particularly in heavily pretreated or frail populations [67,68,69,70]. In combination regimens, increased rates of lymphopenia and venous thromboembolism have been reported, while rare but serious risks of myelodysplastic syndrome and acute myeloid leukemia necessitate long-term hematologic surveillance [67,68,69].
Despite robust efficacy in selected populations, acquired resistance remains a major limitation to durable disease control. Identified mechanisms include BRCA1/2 reversion mutations, replication-fork stabilization, altered PARP expression, drug efflux, and adaptive DNA-damage signaling networks, driving ongoing efforts to optimize sequencing and develop rational combination strategies to minimize cross-resistance [48,60]. Accordingly, current guidelines integrate PARP inhibitors into biomarker-driven treatment algorithms while emphasizing practical challenges related to genomic testing access, toxicity management, and sequencing relative to other life-prolonging therapies [61,71].
The clinical development of olaparib exemplifies precision oncology in prostate cancer: a therapy whose chemically defined mechanism targets a specific molecular vulnerability, whose advancement progressed from exploratory biomarker-unselected studies to definitive randomized trials, and whose continued evolution—through refined diagnostics, combination strategies, and resistance-focused research—continues to shape real-world treatment paradigms for advanced disease [51,52,53,54]. Table 1 summarizes treatment-emergent adverse events associated with olaparib and recommended management strategies; Table 2 presents key pivotal clinical trials evaluating its efficacy and safety; and Figure 3 illustrates comparative rFPS and OS outcomes for olaparib across prostate cancer disease states.
3. Lutetium (177Lu) Vipivotide Tetraxetan
Lutetium (^177^Lu) vipivotide tetraxetan (also known as ^177^Lu-PSMA-617, marketed as Pluvicto) is a radioligand therapeutic agent that delivers targeted β-radiation to cells expressing the transmembrane protein PSMA, which is highly overexpressed on the surface of most prostate cancer cells but has limited expression in normal tissues, thereby enabling selective tumor targeting with relative sparing of nonmalignant organs [82,83,84]. This biologic specificity has made ^177^Lu-PSMA-617 a cornerstone of PSMA-directed “theranostic” strategies and underscores the central importance of accurate biomarker-based patient selection in routine clinical practice.
Following intravenous administration, the PSMA-binding ligand vipivotide tetraxetan binds with high affinity to PSMA on prostate cancer cells, undergoes internalization, and delivers the conjugated radioisotope Lutetium-177, whose emitted β-particles induce DNA damage—particularly double-strand breaks—leading to tumor cell death. The relatively short tissue path length of β-particles enables a beneficial “cross-fire” effect, whereby adjacent tumor cells with lower or heterogeneous PSMA expression may also receive cytotoxic radiation, partially mitigating intratumoral heterogeneity [85,86,87,88]. This mechanism distinguishes RLT from both conventional systemic treatments and external-beam radiotherapy, allowing repeated systemic administration with molecularly targeted cytotoxicity (Figure 4) [89,90,91]. However, heterogeneity of PSMA expression across and within metastatic lesions remains a clinically relevant limitation, influencing both response durability and resistance.
The clinical development and regulatory approval of lutetium (^177^Lu) vipivotide tetraxetan (Pluvicto^®^) represent a major advance in PSMA-directed radioligand therapy (RLT) for mCRPC. The evidence base supporting PSMA-targeted RLT has been built on progressively refined patient selection, imaging-based eligibility criteria, and robust efficacy endpoints. Early phase II and single-arm studies established proof of concept by enrolling PSMA-positive mCRPC patients and demonstrating meaningful PSA responses with manageable toxicity, thereby providing the biological and clinical rationale for further development and directly informing the design of subsequent randomized trials incorporating stringent molecular imaging selection [21,22].
The first major regulatory milestone for PSMA–targeted radioligand therapy occurred on 23 March 2022, when the U.S. Food and Drug Administration (FDA) approved ^177^Lu-PSMA-617 for adult patients with PSMA-positive mCRPC who had progressed following androgen receptor pathway inhibitor (ARPI) therapy and taxane-based chemotherapy. This approval was based on the phase III VISION trial, which mandated PSMA positivity on PET imaging and excluded patients with discordant PSMA-negative/FDG-positive disease, thereby ensuring molecularly appropriate patient selection [93,94,95,96]. In VISION, treatment with ^177^Lu-PSMA-617 plus best standard of care significantly improved both rPFS and OS compared with standard of care alone. Median rPFS increased from 3.4 to 8.7 months (HR 0.40; 95% CI: 0.29–0.56), while median OS improved from 11.3 to 15.3 months (HR 0.62; 95% CI: 0.52–0.74), with consistent benefit across secondary endpoints including objective response and time to symptomatic skeletal events [21,85,87,97]. Importantly, these survival gains were achieved without deterioration in quality-of-life metrics, despite higher rates of grade ≥3 adverse events in the treatment arm [85,98,99].
Additional trials further refined patient selection and comparative efficacy. The randomized phase II TheraP trial mandated dual-tracer PSMA and FDG PET imaging and compared ^177^Lu-PSMA-617 with cabazitaxel, using PSA response as the primary endpoint. TheraP demonstrated superior biochemical response rates and a more favorable toxicity profile for radioligand therapy, reinforcing the critical role of imaging-based enrichment strategies and establishing PSMA PET as an essential biomarker for treatment eligibility and outcome optimization [22]. The therapeutic positioning of ^177^Lu-PSMA-617 has continued to evolve toward earlier disease settings. In the phase III PSMAfore trial, conducted in taxane-naïve patients with PSMA-positive mCRPC who had progressed on a single prior ARPI, radioligand therapy significantly prolonged rPFS compared with switching to an alternative AR-targeted agent, with an acceptable safety profile [100,101,102,103]. These findings led to FDA expansion of the U.S. indication on 28 March 2025 to include patients appropriate for delaying taxane-based chemotherapy [104]. In contrast, regulatory approvals remain region specific: the European Medicines Agency granted marketing authorization on 9 December 2022 for post-taxane mCRPC, while pre-taxane use remains outside the current European label [105]. Real-world evidence has reinforced the external validity of these findings, with retrospective analyses of heavily pretreated mCRPC populations treated outside clinical trials demonstrating PSA response rates comparable to those observed in VISION, supporting the feasibility and effectiveness of PSMA-directed therapy in routine oncology practice [105,106].
Pharmacokinetic, biodistribution, and dosimetry studies—including dedicated substudies within VISION—have provided critical insights for clinical implementation. Following intravenous administration, ^177^Lu-PSMA-617 rapidly distributes to PSMA-expressing tumors and to physiologic uptake sites including kidneys, liver, salivary and lacrimal glands, bladder wall, bone marrow, gastrointestinal tract, and other organs, and is predominantly cleared via the renal route with bi-exponential clearance kinetics. Reported effective half-lives include an initial component of approximately 1.7 ± 0.8 h and a terminal half-life of 41.1 ± 9.3 h [107,108,109]. Organs receiving the highest absorbed radiation doses include the salivary glands and kidneys, as well as the bladder wall, large intestine, lacrimal glands, and rectum, thereby defining dose-limiting organs and informing radiation safety planning and cycle limits [107,108,109]. These dosimetry findings also support the use of PSMA PET–based intensity thresholds for eligibility, as applied in VISION and PSMAfore, where sufficient tumor uptake relative to reference organs was required to ensure favorable tumor-to-organ dose ratios. Because renal handling is central to clearance, renal function and cumulative radiation exposure remain key considerations in patient selection and treatment planning [108,110].
Across clinical trials and post-marketing experience, the safety profile of ^177^Lu-PSMA-617 is characterized by a toxicity spectrum distinct from that of systemic hormonal or targeted therapies. The most common adverse reactions (≥20% of treated patients) include fatigue, xerostomia, nausea, anemia, decreased appetite, and constipation, while the most frequent laboratory abnormalities (≥30%) include lymphopenia, reductions in hemoglobin, leukocytes, and platelets, as well as decreases in serum calcium and sodium [82,85]. In VISION and PSMAfore, grade ≥3 adverse events were reported in approximately 50% of treated patients [100,101,102], and in VISION specifically, grade 3–4 adverse events occurred in approximately 52.7% of patients receiving ^177^Lu-PSMA-617 versus 38.0% in the control arm, although patient-reported quality-of-life measures were largely preserved [85]. Xerostomia and salivary gland toxicity are characteristic adverse effects, occurring in approximately 30–60% of patients but predominantly grade 1–2 and infrequently dose-limiting [21,82,85,111]. In contrast, bone marrow suppression constitutes the principal severe toxicity, with grade ≥3 anemia observed in approximately 10–13% of patients, alongside thrombocytopenia and neutropenia, particularly in individuals with extensive osseous metastatic burden [21,85,105,111]. These observations emphasize careful patient selection, baseline marrow reserve assessment, and long-term hematologic surveillance.
Because ^177^Lu-PSMA-617 is a radiopharmaceutical, cumulative radiation exposure introduces long-term considerations, including renal toxicity, sustained marrow suppression, embryo–fetal toxicity, potential fertility impairment, and theoretical genotoxic, carcinogenic, or secondary malignancy risks, all of which require counseling and longitudinal monitoring [112,113]. Dosimetry analyses further indicate that patients with impaired renal function may receive up to a two-fold higher absorbed kidney dose compared with those with normal creatinine clearance, increasing the likelihood of approaching renal safety thresholds after multiple cycles; accordingly, baseline renal function and bone marrow reserve cut-offs are emphasized as critical eligibility criteria to mitigate nephrotoxicity and cumulative marrow suppression [108,110]. In addition, discordant FDG-positive/PSMA-negative lesions—excluded in VISION and linked to inferior outcomes—have emerged as a clinically meaningful modifier of treatment suitability, reflecting biologically aggressive PSMA-non-avid disease unlikely to benefit from PSMA-targeted radioligand therapy and reinforcing the value of dual-tracer imaging for contemporary patient selection [93,94,95,96].
Despite its targeted mechanism, resistance to ^177^Lu-PSMA-617 can emerge through loss or downregulation of PSMA expression, selection of PSMA-negative tumor clones, limited radiation delivery to bulky or poorly perfused lesions, or intact DNA-damage repair capacity, underscoring the need for rational sequencing and combination strategies to maximize durable benefit. Clinically, lutetium (^177^Lu) vipivotide tetraxetan represents a paradigm shift in prostate cancer management by integrating molecular imaging with targeted systemic radiotherapy, embodying a theranostic approach that uses PSMA PET for patient selection and targeted radiotherapy for disease control [86,89,113,114]. Its expanding role reflects not only demonstrated survival benefit but also its potential to delay chemotherapy or serve as a therapeutic bridge between hormonal therapy and cytotoxic regimens in appropriately selected patients [115,116,117,118]. Ongoing trials are exploring earlier disease settings, optimized dosing and fractionation schedules, and combination approaches with AR blockade, PARP inhibitors, immunotherapy, and chemotherapy [119,120,121,122]. Emerging predictive dosimetry strategies—including physiologically based pharmacokinetic modeling and machine-learning–assisted dose optimization—may further refine patient selection and improve the therapeutic index, particularly in patients with heterogeneous tumor burden or compromised organ function [119,123,124].
^177^Lu-PSMA-617 is therefore a PSMA-targeted radioligand therapy whose development has progressed from early proof-of-concept studies to definitive randomized trials and region-specific regulatory approvals, establishing it as a central component of precision oncology for mCRPC. Continued optimization will depend on refined imaging-based selection, individualized dosimetry, strategic sequencing with other life-prolonging therapies, and deeper understanding of resistance mechanisms. Table 3 summarizes treatment-emergent adverse events and management strategies; Table 4 highlights pivotal clinical trials evaluating efficacy and safety; and Figure 5 shows comparative rPFS and OS outcomes for ^177^Lu-PSMA-617 across prostate cancer disease states.
4. Abiraterone
Abiraterone acetate is an orally administered, selective, irreversible inhibitor of cytochrome P450 17α-hydroxylase/C17,20-lyase (CYP17A1), a critical enzyme in androgen biosynthesis, and is therefore classified as an androgen-biosynthesis–blocking androgen-receptor pathway inhibitor that suppresses extragonadal, intratumoral, and testicular androgen production beyond that achieved with conventional gonadal suppression alone [134,135,136]. By targeting CYP17A1 at a key steroidogenic branch point, abiraterone effectively inhibits the synthesis of dehydroepiandrosterone and androstenedione, the principal precursors of testosterone and dihydrotestosterone (DHT). This mechanism enables sustained androgen deprivation even in castrate conditions, where residual androgen production continues to drive tumor progression (Figure 6) [137,138,139]. In real-world clinical practice, this profound androgen suppression underpins abiraterone’s broad applicability across multiple disease states but also shapes considerations regarding sequencing with other androgen-receptor–directed therapies.
Pharmacologically, abiraterone blocks the conversion of pregnenolone and progesterone into their 17α-hydroxylated derivatives and downstream androgen precursors, resulting in marked reductions in serum testosterone levels in men with castration-resistant prostate cancer (CRPC) [140,141,142]. Preclinical enzymology studies first demonstrated near-complete abrogation of androgen synthesis with selective CYP17A1 inhibition, and early-phase clinical investigations by Attard and colleagues subsequently confirmed that this biochemical suppression translated into clinically meaningful PSA declines in heavily pretreated CRPC patients [143,144,145]. These foundational data established both the biological rationale and clinical feasibility of sustained androgen biosynthesis inhibition.
The clinical evidence supporting abiraterone across the prostate cancer disease continuum is derived from rigorously designed trials with clearly defined patient populations and survival-focused endpoints. Early phase II single-arm studies in mCRPC enrolled heterogeneous cohorts with variable prior therapy exposure and demonstrated consistent PSA and radiographic responses, establishing proof of activity and tolerability in both pre- and post-chemotherapy settings and directly informing the design of subsequent phase III trials with more stringent eligibility criteria and clinically meaningful primary endpoints [26,27].
The first pivotal randomized evidence supporting abiraterone’s survival benefit emerged from the COU-AA-301 trial, a global phase III study conducted in men with post-docetaxel mCRPC. In this setting, abiraterone acetate plus prednisone significantly improved OS, with a median OS of 15.8 months compared with 11.2 months for placebo (absolute gain 4.6 months; HR 0.74; 95% CI: 0.64–0.86), and also prolonged rPFS (median 5.6 vs. 3.6 months; HR 0.66; 95% CI: 0.58–0.76). Additional benefits included higher PSA response rates, improved pain control, and delayed radiographic progression, leading to regulatory approval and establishing abiraterone as a standard post-chemotherapy option [146,147,148].
This benefit was subsequently extended to chemotherapy-naïve mCRPC in the COU-AA-302 trial, a randomized, double-blind phase III study with co-primary endpoints of OS and rPFS. Abiraterone plus prednisone prolonged median OS to 34.7 months compared with 30.3 months for placebo (absolute gain 4.4 months; HR 0.81; 95% CI: 0.70–0.93) and significantly delayed radiographic progression (HR 0.43; 95% CI: 0.35–0.52), while also extending time to opiate use and preserving patient-reported quality of life [149,150,151]. Together, these findings confirmed that androgen biosynthesis remains a clinically relevant therapeutic target even after the development of castration resistance.
Patient selection was further broadened to earlier disease states in the LATITUDE and STAMPEDE trials. LATITUDE demonstrated that adding abiraterone to androgen deprivation therapy (ADT) in patients with high-risk metastatic hormone-sensitive prostate cancer significantly improved rPFS (30.7 vs. 18.3 months; HR 0.53; 95% CI: 0.37–0.76) and OS (HR 0.62; 95% CI: 0.51–0.76), yielding substantial absolute survival gains [152,153,154,155]. Similarly, the multi-arm STAMPEDE platform trial showed that intensification of ADT with abiraterone significantly improved OS compared with ADT alone, with hazard ratios ranging from approximately 0.62 to 0.72 and absolute median OS gains of roughly 12–16 months in metastatic subgroups [29]. Subsequent analyses confirmed consistent benefit across both low- and high-volume metastatic disease, reinforcing abiraterone’s versatility and informing real-world treatment algorithms across diverse patient populations [156,157,158,159].
Real-world evidence has corroborated these trial results, demonstrating comparable outcomes in broader and more heterogeneous populations, including older patients and those with cardiovascular comorbidities or extensive prior therapy [160,161,162]. Translational studies have identified biomarkers that may refine patient selection and sequencing decisions, most notably the AR splice variant AR-V7 detected in circulating tumor cells, which has been associated with reduced responsiveness to abiraterone. These observations support biomarker-driven stratification to guide transitions to alternative treatment classes such as taxanes or PSMA-targeted radioligand therapy, although AR-V7 testing is not yet routinely implemented [160,161,162].
Abiraterone’s safety profile reflects its mechanism of CYP17-mediated adrenal steroidogenesis inhibition. Suppression of cortisol synthesis results in compensatory adrenocorticotropic hormone elevation and mineralocorticoid excess, manifesting clinically as hypertension, hypokalemia, and fluid retention, with occasional cardiac arrhythmias. Across pivotal trials including COU-AA-301, COU-AA-302, and LATITUDE, grade ≥3 adverse events were reported in approximately 40–55% of patients [26,27,28]. Grade ≥3 hypertension occurred in approximately 10–20% of patients, while grade ≥3 hypokalemia was observed in 5–10%; fluid retention and edema were common but predominantly low grade [163,164,165]. These toxicities typically occurred early during treatment and were generally manageable with routine coadministration of corticosteroids, antihypertensive therapy, electrolyte monitoring, and dose modification when required. Hepatic enzyme elevations, fatigue, and infrequent but clinically significant hepatotoxicity necessitate regular laboratory surveillance [166,167]. Cardiovascular toxicity remains a key consideration, particularly in patients with preexisting cardiovascular disease, and often influences treatment selection and sequencing when alternative androgen receptor–directed or non-hormonal therapies are available [168,169].
Despite its established efficacy, resistance to abiraterone inevitably develops. Mechanisms include AR amplification or mutation, intratumoral androgen synthesis bypassing CYP17 inhibition, activation of alternative steroidogenic pathways, and emergence of constitutively active AR splice variants such as AR-V7. These resistance mechanisms limit response durability and contribute to cross-resistance with other androgen receptor–directed therapies, underscoring the importance of timely transition to mechanistically distinct treatments such as chemotherapy, PARP inhibitors, or PSMA-targeted radioligand therapy [170,171,172,173].
Combination strategies have therefore been extensively explored. Abiraterone has been evaluated in combination with PARP inhibitors, next-generation AR antagonists, radiopharmaceuticals, and immunotherapeutic agents, with preclinical and early clinical data suggesting potential synergy through enhanced DNA-damage susceptibility and modulation of the tumor microenvironment [170,171,172,173,174,175]. However, negative results from trials such as CYCLONE 2, which failed to demonstrate benefit from adding the CDK4/6 inhibitor abemaciclib to abiraterone in an unselected mCRPC population, highlight the necessity of strong biological rationale and appropriate patient selection when extending combination approaches [31].
Collectively, these studies illustrate how differences in trial design, patient population, biomarker integration, and endpoint selection have shaped the optimal clinical positioning and sequencing of abiraterone-based therapy. Abiraterone acetate exemplifies mechanism-driven treatment in advanced prostate cancer, with extensive validation across hormone-sensitive and castration-resistant settings and continued relevance as a therapeutic backbone within evolving multimodal strategies. As treatment landscapes increasingly incorporate PARP inhibitors, PSMA-targeted radioligand therapy, and next-generation androgen receptor blockade, abiraterone’s role will continue to be refined through biomarker-driven patient selection, rational sequencing, and resistance-aware treatment strategies [176,177,178,179].
Intensification of AR signaling inhibition with abiraterone confers substantial survival benefit across metastatic hormone-sensitive and castration-resistant prostate cancer, with hazard ratios ranging from approximately 0.62 to 0.81 and absolute median OS gains of roughly 4–22 months depending on disease setting and baseline risk [26,27,28]. Abiraterone remains a cornerstone of contemporary precision oncology for prostate cancer, supported by a well-characterized mechanism, robust survival benefit, and a manageable safety profile. Ongoing research continues to optimize sequencing, overcome resistance, and extend benefit to a broader range of patients. Table 5 summarizes treatment-emergent adverse events and recommended management strategies; Table 6 highlights key pivotal clinical trials assessing efficacy and safety; and Figure 7 presents comparative rPFS and OS outcomes for abiraterone across prostate cancer disease states.
5. Anticipated Developments
Future therapeutic strategies in prostate cancer increasingly emphasize precision oncology, convergence of biological pathways, and multimodal integration, reflecting a fundamental shift toward treatments that are mechanistically informed yet dynamically adaptable to tumor evolution. Advances in PARP inhibition, PSMA-targeted RLT, and next-generation androgen-receptor (AR) blockade now constitute the conceptual and clinical backbone of emerging therapeutic frameworks, each grounded in a deeper understanding of resistance biology and disease heterogeneity [184,185]. Despite these advances, their real-world implementation remains constrained by important limitations related to toxicity burden, access to specialized diagnostics and infrastructure, financial cost, and persistent gaps in clinical evidence, underscoring the need for continued critical evaluation alongside innovation.
Expanding translational research efforts are increasingly focused on linking molecular phenotypes with therapeutic vulnerabilities to enable rational combinations across treatment classes. For example, ongoing work suggests that combined PARP inhibition and androgen-signaling suppression may ultimately extend beyond canonical BRCA1/2- or HRR-mutant populations. This evolution is driven by increasingly sophisticated genomic, epigenomic, and functional biomarker analyses that define synthetic-lethal interactions with greater granularity [186,187]. However, widespread clinical adoption of such approaches is currently limited by the lack of standardized assays, variable reproducibility across laboratories, and uncertainty regarding how best to integrate emerging biomarkers into routine treatment algorithms.
Novel biomarker platforms—including genomic scarring signatures that reflect cumulative homologous recombination deficiency, RAD51 foci assays that provide functional readouts of HRR activity, and circulating tumor DNA profiling that captures intrapatient heterogeneity over time—offer promising tools for more precise patient stratification [188,189,190,191,192,193,194]. While these technologies hold significant potential to expand the therapeutic reach of agents such as olaparib, they also raise unresolved questions regarding cost-effectiveness, regulatory validation, and optimal thresholds for clinical decision-making. Prospective trials incorporating these biomarkers as stratification or enrichment tools will be essential to determine their true clinical utility.
Radioligand therapy, including Lutetium (^177^Lu) vipivotide tetraxetan, represents one of the most transformative advances in metastatic prostate cancer management, with compelling evidence of PSA responses, radiographic improvement, and survival benefit even in heavily pretreated disease. Nonetheless, its broader adoption is limited by logistical complexity, dependence on PSMA PET imaging, radiation-safety infrastructure requirements, and variable global availability. Current trials exploring earlier use of RLT and combinations with hormonal intensification or DNA repair–modulating agents are biologically compelling, particularly given largely non-overlapping toxicity profiles and potential synergy through immune activation and amplified DNA damage [195,196,197,198,199]. However, unanswered questions remain regarding cumulative radiation exposure, optimal dose fractionation, long-term renal and marrow toxicity, and cost sustainability—issues that must be addressed before widespread front-line implementation can be justified.
Next-generation AR pathway inhibitors and rational combination strategies remain central to both current practice and future innovation, driven by detailed characterization of resistance mechanisms such as AR amplification, constitutively active splice variants, epigenetically driven lineage plasticity, and neuroendocrine transdifferentiation. In response, novel therapeutic classes—including PROTAC-based AR degraders, N-terminal domain inhibitors, and dual-pathway modulators—have been developed to maintain AR suppression despite complex molecular adaptations [4,200,201,202]. While these approaches are conceptually attractive, their long-term safety, sequencing relative to established agents, and potential for cross-resistance remain incompletely understood, highlighting the need for carefully designed comparative and biomarker-enriched clinical trials.
The integration of radiomics, artificial-intelligence–driven treatment selection, and adaptive therapy principles offers a potential pathway to overcoming many current limitations. AI-enhanced PSMA PET quantification, automated risk-stratification models, and machine-learning algorithms trained on multimodal clinical datasets may improve prediction of response, refine sequencing decisions, and reduce overtreatment [4,203,204,205]. However, these technologies introduce new challenges related to data standardization, interpretability, regulatory oversight, and equitable access, emphasizing that technological sophistication must be matched by pragmatic implementation strategies.
Immunotherapy remains an area of high unmet need in prostate cancer. Although historical efficacy has been modest, emerging approaches combining checkpoint blockade with radioligand-induced immunogenic cell death, PARP inhibitor–mediated STING activation, and modulation of myeloid-driven immune suppression have shown early promise [205,206,207,208]. The heterogeneity of observed responses underscores the importance of personalized immune profiling, yet such approaches are currently resource-intensive and not standardized, limiting near-term applicability. Future research must clarify which immune phenotypes are most likely to benefit and how immunotherapy can be rationally sequenced or combined without excessive toxicity.
Parallel advances in understanding metabolic vulnerabilities—including alterations in cholesterol synthesis, mitochondrial metabolism, oxidative stress pathways, and DNA repair–associated metabolic dependencies—have opened additional therapeutic avenues. Targeting enzymes such as AKR1C3, fatty acid synthase, and regulators of glutamine metabolism has shown potential to sensitize tumors to existing treatments and suppress aggressive phenotypes in resistant disease models [209,210,211]. Translating these findings into clinically viable therapies will require overcoming challenges related to systemic toxicity, metabolic redundancy, and patient selection.
Liquid biopsy technologies are poised to play a central role in addressing many unresolved clinical questions. Serial analysis of circulating tumor cells, ctDNA mutational profiles, and methylation signatures offers a noninvasive means of monitoring tumor evolution, detecting resistance early, and guiding adaptive treatment strategies [212,213,214]. While these approaches promise to reduce unnecessary toxicity and enable more dynamic care, their integration into routine practice will depend on prospective validation, cost containment, and clear demonstration of outcome benefit.
Ultimately, the convergence of precision radiotherapy, targeted systemic agents, and advanced molecular diagnostics is likely to define the next phase of prostate cancer management. Increasingly individualized treatment pathways will rely on multidisciplinary collaboration across genetic counseling, molecular pathology, nuclear medicine, urologic oncology, medical oncology, and radiation oncology. This evolution reflects a broader transition toward biology-guided medicine, in which therapeutic decisions are informed less by conventional staging and more by real-time molecular, functional, and imaging biomarkers [215,216,217]. Future research must therefore prioritize not only therapeutic innovation but also implementation science, health equity, and long-term survivorship considerations to ensure that advances in precision oncology translate into meaningful, durable benefit for patients. Table 7 presents contemporary management strategies for prostate cancer, outlining current treatment approaches, while Figure 8 illustrates therapeutic pathways and treatment algorithms in mCRPC, integrating biomarkers and treatment sequencing.
6. Conclusions
Emerging therapeutic strategies in prostate cancer reflect a rapidly evolving landscape in which molecular profiling, precision radioligand targeting, and next-generation hormonal manipulation are redefining standards of care. The integration of PARP inhibition with agents such as olaparib, PSMA-targeted radioligand therapy using lutetium (^177^Lu) vipivotide tetraxetan, and potent androgen-receptor pathway suppression with abiraterone has demonstrated that maximal therapeutic benefit is achieved when complementary biological vulnerabilities are addressed concurrently. Together, these modalities underscore a transition from uniform treatment paradigms toward individualized, biology-driven algorithms guided by genomic alterations, functional imaging, and tumor-specific characteristics. Across multiple clinical investigations, both combination and sequential strategies have yielded meaningful improvements in rPFS, biochemical response rates, and OS while maintaining acceptable safety profiles. Despite these advances, significant challenges persist, including the development of acquired resistance, the need for more precise and predictive biomarkers of response, and unresolved questions regarding optimal treatment sequencing to prolong disease control. Future research will increasingly prioritize rational combination strategies, earlier implementation of targeted therapies in hormone-sensitive disease, and refinement of theranostic approaches that integrate molecular imaging with selective delivery of therapeutic payloads. As the evidence base continues to expand, these innovations are expected to converge into comprehensive, biology-driven treatment frameworks that broaden patient eligibility, enhance the depth and durability of response, and ultimately improve long-term outcomes for individuals with advanced prostate cancer.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Sung H. Ferlay J. Siegel R.L. Laversanne M. Soerjomataram I. Jemal A. Bray F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries CA Cancer J. Clin.20217120924910.3322/caac.2166033538338 · doi ↗ · pubmed ↗
- 2Siegel R.L. Giaquinto A.N. Jemal A. Cancer Statistics, 2024 CA Cancer J. Clin.202474124910.3322/caac.2182038230766 · doi ↗ · pubmed ↗
- 3Sandhu S. Moore C.M. Chiong E. Beltran H. Bristow R.G. Williams S.G. Prostate cancer Lancet 20213981075109010.1016/S 0140-6736(21)00950-834370973 · doi ↗ · pubmed ↗
- 4Watson P.A. Arora V.K. Sawyers C.L. Emerging Mechanisms of Resistance to Androgen Receptor Inhibitors in Prostate Cancer Nat. Rev. Cancer 20151570171110.1038/nrc 401626563462 PMC 4771416 · doi ↗ · pubmed ↗
- 5Abida W. Armenia J. Gopalan A. Brennan R. Walsh M. Barron D. Danila D. Rathkopf D. Morris M. Slovin S. Prospective Genomic Profiling of Prostate Cancer Across Disease States Reveals Germline and Somatic Alterations That May Affect Clinical Decision Making JCO Precis. Oncol.20172017 PO.17.0002910.1200/PO.17.0002928825054 PMC 5558263 · doi ↗ · pubmed ↗
- 6Eiber M. Fendler W.P. Rowe S.P. Calais J. Hofman M.S. Maurer T. Schwarzenboeck S.M. Kratowchil C. Herrmann K. Giesel F.L. Prostate-Specific Membrane Antigen Ligands for Imaging and Therapy J. Nucl. Med.20175867 S 76S 10.2967/jnumed.116.18676728864615 · doi ↗ · pubmed ↗
- 7Sands M. Adams S. Lee J. Li M. Wang M. Walsh T.Jr. Leon L. Zablah A. Haerens M. Liu Z. The Interconnection between Androgen Receptor and DNA Damage Response Pathways in Prostate Cancer Curr. Urol.20251937638710.1097/CU 9.000000000000030041058763 PMC 12499796 · doi ↗ · pubmed ↗
- 8Leão R. Domingos C. Figueiredo A. Hamilton R. Tabori U. Castelo-Branco P. Cancer Stem Cells in Prostate Cancer: Implications for Targeted Therapy Urol. Int.20179912513610.1159/00045516028142149 · doi ↗ · pubmed ↗