Heritability and Transcriptional Impact of JAK3, STAT5A and STAT6 Variants in a Tyrolean Family
Hye Kyung Lee, Teemu Haikarainen, Yasemin Caf, Priscilla A. Furth, Ludwig Knabl, Olli Silvennoinen, Lothar Hennighausen

TL;DR
This study explores how rare genetic variants in immune-related genes affect gene activity in a family without causing obvious health issues.
Contribution
The study identifies germline variants in JAK3, STAT5A, and STAT6 and investigates their transcriptional effects in a family without clinical symptoms.
Findings
Rare JAK3P151R, JAK3R925S, STAT5AV494L, and STAT6Q633H variants were found in a three-generation family.
Not all carriers of these variants showed the same immune transcriptome activation.
Germline variants may have context-dependent effects influenced by other genetic and environmental factors.
Abstract
The Janus Kinase (JAK) and Signal Transducers and Activators of Transcription (STAT) pathways regulate a range of biological processes, including immune response and hematopoiesis. While a major research focus has been on somatic human mutations in disease, less is known about the heritability of germline variants and their physiological impact. This study addresses an important issue in population genetics: the context-dependent effects and incomplete penetrance of rare genetic variants in immune pathways. Here we identify the rare JAK3P151R, JAK3R925S, STAT5AV494L, and STAT6Q633H variants in an extended family spanning three generations, integrate in silico analyses and AlphaFold 3 structural predictions, and investigate the immune transcriptomes in probands carrying one or more variants. All four variants are inherited through the germline without any evident clinical or…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
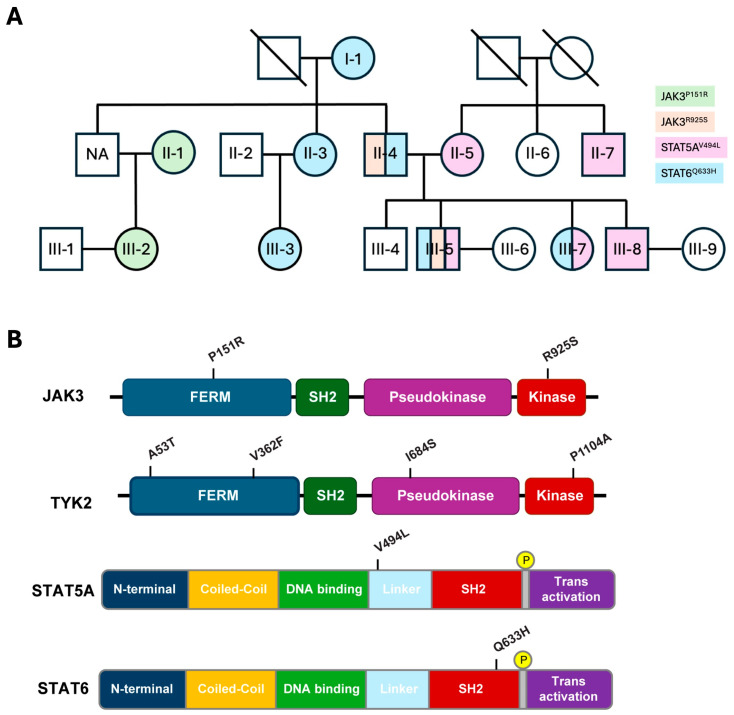 Figure 1
Figure 1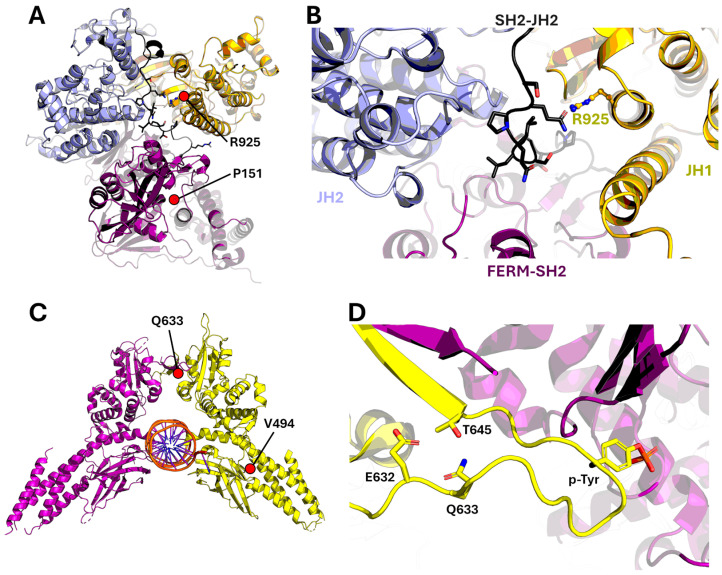 Figure 2
Figure 2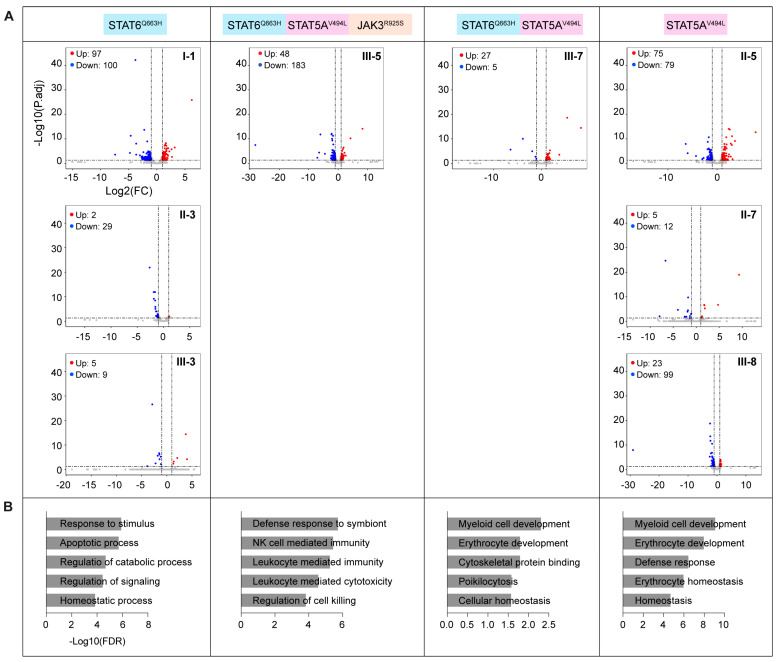 Figure 3
Figure 3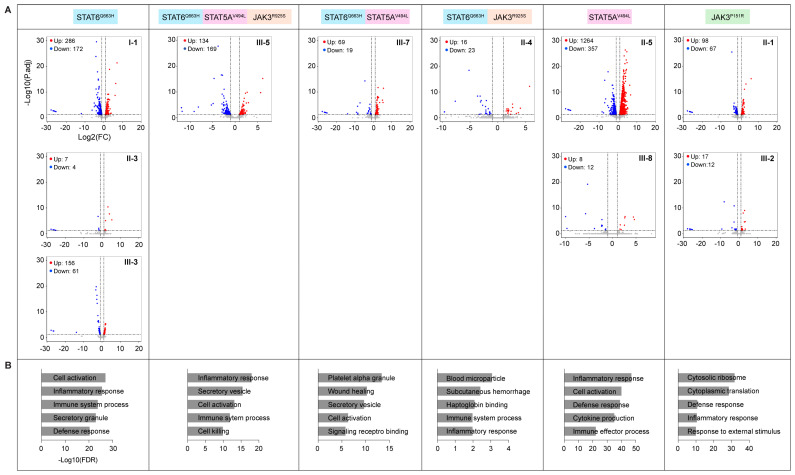 Figure 4
Figure 4- —National Institutes of Health (NIH)
- —Research Council of Finland
- —Sigrid Juselius Foundation
- —Finnish Cancer Foundation
- —Tampere Tuberculosis Foundation
- —Tampere University Hospital
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMyeloproliferative Neoplasms: Diagnosis and Treatment · Cytokine Signaling Pathways and Interactions · Immunodeficiency and Autoimmune Disorders
1. Introduction
Janus Kinases (JAKs) and Signal Transducers and Activators of Transcription (STATs) play indispensable roles in translating cytokine signals into transcriptional responses that regulate a range of biological processes, including body growth [1], lactation [2], hematopoiesis [2,3,4], inflammation, and antiviral defense. Dysregulated JAK/STAT signaling has been implicated in chronic inflammation and autoimmune disease, and somatic variants have been identified predominantly in hematological malignancies. These include the classical JAK2^V617F^ variant in myeloproliferative neoplasms (MPNs) [5,6] and Src Homology 2 (SH2) domain variants of STAT family members [7,8].
We recently identified a range of germline JAK/STAT variants and linked some to an exacerbated innate immune response after vaccination [9,10] and an elevated baseline immune transcriptome [11]. Access to community-based cohorts permitted us to address yet unanswered questions focused on the presence of JAK/STAT variants in large families. First, it is not well understood to what extent these variants are transmitted through the germline, especially in individuals carrying two or more variants. Second, the combinatorial transcriptional impact of several variants in individuals remains to be understood. Here we identified a large family carrying two JAK3 variants (JAK3^P151R^ and JAK3^R925S^), as well as STAT5A^V494L^ and STAT6^Q633H^ variants. These germline variants are very rare but no definitive biological information has been reported in the literature [12]. The JAK3^P152R^ variant results in a non-conservative amino acid change in the FERM domain and the JAK3^R925S^ variant is also a non-conservative amino acid change in the kinase domain. STAT5A^V494L^, a novel germline variant, represents a conservative substitution, located in a solvent-exposed loop at the end of the DNA binding domain of the protein. The STAT6^Q633H^ variant is located in the SH2 linker region and gain.
The novel conceptual contribution of our study extends beyond the report-of-function (GOF) germline mutations have been identified in this region [13]. We used in silico analyses and AlphaFold 3 (AF3) protein structure predictions to assess the pathogenicity of these variants and gauged their impact in individuals carrying one or more variants.ing of rare JAK/STAT germline variants in a large family and their inheritance through three generations to understanding how these variants could shape the immune transcriptome. While this concept is tenable, the low frequency of these variants makes it difficult, if not impossible, to identify additional individuals displaying these variant combinations. This highlights the possibility that combinations of JAK/STAT variants have an individual impact. However, this complexity is further expanded by including variants in Tyrosine Kinase 2 (TYK2) and additional proteins acting upstream of JAK/STAT, such as receptors, and downstream, such as SOCS proteins. Just by including TYK2 variants, each of the investigated family members has a unique combination of variants. Lastly, while AF3 is a powerful tool in protein modeling, it might not provide sufficient depth to identify subtle changes leading to protein dimerization, receptor–JAK-STAT interactions, STAT nuclear translocation, and transcriptional activation. Structural neutrality, as shown in some of our variants, does not necessarily equate to a lack of biological impact.
2. Results
In a quest to understand the heritability of JAK/STAT variants and further explore their combined transcriptional impact, we searched for families carrying two or more variants in any of the three JAK and seven STAT proteins. Here we report on a family we had recruited during the COVID-19 pandemic to investigate the innate immune response after COVID-19 Omicron infection [14]. We chose this family for a more detailed investigation of the influence of common and rare variants in the JAK/STAT signaling pathway on the immune transcriptome. Importantly, members of three generations of this family were examined; all family members lived in the same geographical area and had similar lifestyles. This enabled us to make more precise statements about the significance of the rare variants we found, as environmental or lifestyle-associated confounders could be largely ruled out. Furthermore, a particularly rare variant (STAT6^Q633H^) occurred in members of all three generations, allowing us to assess its potential pathogenicity across different stages of life. We obtained extensive medical data and immune transcriptome data from this family, which was obtained when examining family members during an Omicron infection that affected almost all members at the same time.
Both RNA-seq and whole exome sequencing (WES) data collected from 17 members of a Tyrolean family spanning three generations revealed two JAK3 variants, and one variant each in STAT5A and STAT6 was identified (Figure 1). The JAK3^P151R^ and JAK3^R925S^ variants are in the FERM domain and the kinase domain, respectively. The STAT5A^V494L^ variant is within the linker separating the DNA-binding domain (DBD) and the SH2 domain, and the STAT6^Q633H^ variant is positioned at the C-terminal boundary of the SH2 domain. The STAT6^Q633H^ variant was identified in six individuals in three generations and the STAT5A^V494L^, JAK3^P151R^, and JAK3^R925S^ variants in two generations. Two individuals carried only a single variant, one person carried the STAT6^Q633H^ and JAK3^R925S^ variants, one carried the STAT6^Q633H^ and STAT5A^V494L^ variants, and one person carried three variants, STAT6^Q633H^, STAT5A^V494L^, and JAK3^R925S^ (Table S1). Both JAK3^P151R^ and JAK3^R925S^ are rare variants; STAT5A^V494L^ has not been listed in databases, and STAT6^Q633H^ is an ultra-rare variant with an allele frequency of approximately 10^−4^ in gnomAD and All of Us (Table 1). These findings demonstrate that all four variants are inherited through the germline without any evident clinical or physiological manifestations in the carriers. Importantly, individual II-4 passed on the STAT6^Q633H^ and JAK3^R925S^ variants to his son who also inherited the STAT5A^V494L^ variant from his mother, II-5 (Figure 1). Ten of the eleven family members with JAK/STAT variants also featured TYK2 variants (Table S1). Three family members carried only TYK2 variants, and three family members did not carry any JAK/STAT/TYK2 mutations. Coinheritance of JAK/STAT and TYK2 variants has also been described in other cohorts in South Korea [10] and Austria [9].
To evaluate the potential pathogenicity of these four JAK/STAT variants and the four TYK2 variants, we employed the NIH ClinVar database and additional in silico prediction tools (Table 1). While no ClinVar records were available for STAT6^Q633H^, STAT5A^V494L^ and JAK3^R925S^, JAK3^P151R^ was considered benign. AlphaMissense [15], a state-of-the-art computational tool, predicted an ambiguous score for JAK3^R925S^ and benign scores for the other three variants. The PolyPhen-2 [16] scores predicted a significant impact of the JAK3^R925S^ and STAT6^Q633H^ variants. The REVEL (Rare Exome Variant Ensemble Learner) [17] scores suggested a higher probability of pathogenicity for JAK3^R925S^, STAT5A^V494L^, and STAT6^Q633H^. While the TYK2 variants V362F, I684S, and P1104A are quite common, with an AF between approximately 2 × 10^−1^ and 2 × 10^−2^, the A53T variant has an AF of 7 × 10^−3^. All four are considered benign or likely benign in ClinVar. AlphaMissense and PolyPhen2 categorizes TYK2^I684S^ and TYK2^P1104A^ as pathogenic. TYK2^P1104A^ has also been described as an autoimmune protective variant [18] and suppressor of inflammation [19]. Similarly, TYK2^I684S^ has been reported to protect from psoriasis and impair memory T-cells [20]. To assess potential epistatic effects of the JAK/STAT and TYK2 variants identified in this family, we performed epistasis analyses using the Epistasis Disease Atlas [21] and detected no significant interactions between these variants and other immune-related genes. Maximum-likelihood model scores from NeEDL are summarized in Table S2.
2.1. Structural Analysis of JAK and STAT Mutations
JAK3 has very little structural information available compared to other family members, and only the structure of the JAK3 kinase domain has been experimentally resolved. Therefore, AI-based structure prediction with AF3 was used to obtain structural information of the two identified mutations. Notably, structural prediction of JAK3 with AF3 results in a highly similar tertiary structure when compared to JAK1, JAK2, and TYK2 [18]. We also analyzed the mutations in the context of a cryogenic electron microscopy (cryo-EM) structure of activated JAK dimers (PDB code 8EWY), but in the activated conformation, both mutated residues faced the solvent and did not participate in intra-domain or JAK-JAK interactions across the dimer interface. Therefore, it is likely that the mutations exert their effects, if any, on the monomeric, autoinhibited conformation.
The P151R mutation is located in the FERM-SH2 domain in a short linker between two a-helices (Figure 2A). The residue is solvent-exposed, and based on the structural analysis, the mutation would not disrupt or create any specific interactions. However, due to its rigid cyclic structure, introducing a more flexible residue in the place of proline could lead to disruption of the linker and the conformation of the protein fold, at least locally. The R925S mutation is positioned in the C-lobe of the kinase domain, and in the AF3 model, the arginine side chain faces the SH2-JH2 linker, which runs in the interface of FERM-SH2, pseudokinase, and kinase domains (Figure 2A,B). The mutation could disrupt the linker conformation and thus the autoinhibited conformation of JAK3. Importantly, the SH2-JH2 linker is a hotspot of pathogenic JAK mutation and harbors activating mutations, especially in hematologic malignancies, e.g., JAK3^M511I^ [22] and JAK2^K539L^ [23].
The identified STAT5A^V494L^ mutation is located in a solvent-exposed loop at the end of the DNA binding domain (Figure 2C). Wild-type valine does not participate in any direct interactions via its side chain, which points towards the solvent. Therefore, the conservative Val to Leu mutation is likely benign. STAT6^Q633H^ is in the SH2 domain next to the phosphotyrosine binding site responsible for STAT dimerization (Figure 2C,D). The glutamine residue makes a hydrogen bond with Thr645, contributing to the stability of the phosphotyrosine-containing loop. The Q633H likely conserves this interaction, as histidine can form a similar hydrogen bond via its side chain. No drastic effects are therefore suspected from this mutation.
2.2. STAT5A and STAT6 Variants Are Associated with Enhanced Basal Immune Transcriptomes
To explore the potential impact of these JAK/STAT variants on the baseline immune transcriptome, i.e., in the absence of known infections, in a real-world setting, we conducted RNA-seq on peripheral blood mononuclear cells (PBMC) from 13 family members (Table S3). Specifically, we had baseline transcriptome data from five participants carrying the STAT6^Q633H^ variant. Three members (I-1, II-3, and III-3), spanning three generations carried only the STAT6^Q633H^ variant, while III-7 also carried the STAT5A^V494L^ variant. Individual III-5 carried both the STAT6^Q633H^ and the STAT5A^V494L^ variants as well as the JAK3^R925S^ variant. Volcano plots were generated to visualize differentially expressed genes in family members carrying JAK and/or STAT variants compared with family members without JAK/STAT variants (Figure 3A, Table S3). While a significant number of genes were deregulated in individual I-1 (STAT6^Q633H^ variant), her daughter and granddaughter did not display an aberrant immune transcriptome. The differences observed between these three individuals could be explained be age differences, with I-1 being 87 years old, II-3 55 years old, and III-3 36 years old. Alternatively, additional mutations in immune regulatory pathways could explain the result. Individual III-5 carrying the STAT6^Q633H^ variant in combination with the STAT5A^V494L^ and JAK3^R925S^ variants also presented an elevated transcriptome. Among the individuals carrying the STAT5A^V494L^, II-5 did display an enhanced transcriptome. Gene enrichment analysis of the significantly upregulated genes revealed activation of immune-related pathways, including homeostatic processes, immune response, and myeloid/erythroid cell development, in individuals harboring JAK/STAT variants (Figure 3B, Table S3). The presence of various combinations of [20] variants in these individuals (Table S1) complicates the interpretation of the findings.
We had previously analyzed RNA-seq data from a cohort infected with the SARS-CoV-2 Omicron strain [14], including 13 individuals of the family investigated in this study, and compared differentially expressed genes between individuals carrying JAK and/or STAT variants and family members without JAK/STAT variants. Blood had been collected approximately two weeks after the onset of symptoms and RNA-seq was conducted on PBMCs. Volcano plots demonstrated the highest induction of gene expression in I-1 (STAT6^Q633H^), II-5 (STAT5A^V494L^), III-7 (STAT6^Q633H^; STAT5A^V494L^), III-5 (STAT6^Q633H^; STAT5A^V494L^; JAK3^R925S^), and II-4 (STAT6^Q633H^; JAK3^R925S^) (Figure 4A, Table S4). Notably, individuals with elevated transcriptomes after SARS-CoV-2 Omicron infection also had elevated baseline transcriptomes (Figure 3), suggesting a genetic component, possibly the JAK/STAT variants described here. Across groups carrying distinct JAK and/or STAT variants, significantly upregulated genes were consistently enriched for inflammatory and immune response pathways compared with individuals lacking JAK/STAT mutations (Figure 4B, Table S4).
3. Discussion
The biological function of most germline and somatic human JAK/STAT variants remains elusive, and the impact of combinatorial SNVs and the possible role of epistasis on physiological responses in humans add additional layers of complexity. Such biological questions can be studied best in families carrying combinations of different variants. Here we identify four very rare JAK/STAT variants in a Tyrolean family with 17 members and integrate data-driven approaches and RNA transcriptomes with the goal to further elucidate their functions, alone and in combinations. We demonstrate that all four JAK/STAT variants are inherited in a Mendelian fashion without any evident clinical or physiological manifestations in the carriers. Most notably, only some carriers of specific variants presented with an altered immune transcriptome, suggesting a higher complexity of coinciding JAK/STAT and other immune regulatory SNVs.
With only limited clinical details available of the rare mutations, we employed in silico predictions, which, however, presented a complex and somewhat contradictory picture, with some tools suggesting potential pathogenicity while others indicated benign effects. While AI-driven AF3 is a very powerful tool to predict protein structures, it provided limited insight into the biological impact of the variants under investigation. The inconsistency among computational predictions highlights the difficulty of depending solely on in silico approaches to evaluate how mutations affect complex signaling pathways, emphasizing the importance of experimental confirmation. In vitro experiments, such as investigating JAK activities or chromatin binding of STATs, could provide limited information on individual variants; their combinatorial impact in individuals would be difficult to gauge.
Three variants, JAK3^P151R^, JAK3^R925S^, and STAT6^Q633H^ could be suspected as GOF variants because somatic versions are documented in the Catalog of Somatic Mutations in Cancer (COSMIC). The previous literature documented the presence of nine of the eleven known germline STAT6 GOF variants in COSMIC [13,25,26]. JAK3^R925S^ is a rare variant that has been reported in patients with T-ALL [27], but by itself, it does not transform Ba/F3 cells in vitro or stimulate cell proliferation of the MOHITO cell line [28]. While JAK3^R925S^ has been described as a passenger mutation in T-ALL patients, we suggest that it could impact the basal immune transcriptome and virus-induced innate immune responses in the presence of additional JAK/STAT mutation lines in proband III-5, who also carries the STAT5A^V494L^ and STAT6^Q633H^ variants. A possible synergy between JAK variants has also been reported in leukemia patients carrying two or more variants [29]. STAT6^Q633H^ is a very rare variant that has not yet been reported in the literature. However, one case has been reported in the COSMIC database, and PolyPhen2 reports it as pathogenic. This variant is located at the intersection of the SH2 and TAD domain, in close vicinity of well-known GOF variants, including P643R [13,26] and the key tyrosine phosphorylation site (Y641). Since only one out of the three individuals in our study carrying the STAT6^Q633H^ variant displayed an elevated basal and SARS-CoV-2-induced innate transcriptome, it is likely that additional variants in immune regulators contribute to its function. Like the STAT6 variant, only one of the three individuals carrying the very rare STAT5A^V494L^ displayed an enhanced basal transcriptome, again, suggesting the presence of variants in additional immune regulatory genes. Several hundred variants were identified in these individuals of other key immune genes, such as TYK2 and interferon receptors, and the complexity of variant interactions remains to be investigated. The low AF of some of the mutations makes it unlikely that additional individuals carrying specific combinations of JAK/STAT/TYK2 variants can be found. Only 1 out of 1 million individuals will carry two rare mutations, and some of our study participants carry up to four JAK/STAT/TYK2 variants. This exemplifies the challenges in pinpointing the impact of variants in real-world settings.
Our findings highlight the combinatorial complexity of JAK/STAT variants in humans and their likely impact on physiology and pathophysiology. From a molecular and population genetics perspective, our findings are consistent with context-dependent associations compatible with incomplete penetrance and modifier-gene effects. It also underscores the importance of obtaining WGS data in any family study, permitting the identification of cooperating variants in related pathways. This presence of cooperating variants is highlighted by findings that the MPN driver mutation JAK2^V617F^ has been found in large cohorts of healthy elderly individuals [30]. Since mutations in humans do not exist in isolation, their impact can only be gauged in the context of additional mutations in their native setting. Understanding the complexity of epistatic SNP interactions has become a major challenge [21] in understanding how the co-presence of several variants can modulate physiological and pathophysiological responses. Artificial intelligence-driven algorithms will be critical in integrating the data on individual gene variants and their biological and clinical effects.
Limitations
A limitation of the investigation is that we had identified only one extended family carrying the four JAK/STAT variants. The study depends on in silico predictions, structural modeling, and transcriptomic associations. These methods by themselves cannot determine gain- or loss-of-function effects. Functional in vitro assays could provide some biological information on individual JAK/STAT variants but would not yield significant information on the combinatorial impact of several co-inherited variants. An additional limitation is that the study spanned three generations but each generation contained only limited numbers of individuals to study, therefore we could not explore how age or sex might modify influence of a specific variant’s impact on the basal transcriptome.
4. Materials and Methods
4.1. Study Participants
Single-nucleotide variation (SNV) analyses were performed using 13 publicly available RNA-seq datasets derived from a previously described family infected with the SARS-CoV-2 Omicron variant [14,31]. Whole-exome sequencing (WES) data were available for nine of these individuals, along with six additional family members, yielding a total of 17 participants with SNV data (Figure 1). The cohort comprised nine females (mean age 49 ± 17 years) and eight males (mean age 45 ± 15 years), all of Austrian ethnicity and residing in Tyrol, Austria [14]. Owing to a limited sample size, the influence of sex and/or gender could not be assessed, which limits generalizability.
The study was approved by the Institutional Review Board (IRB) of the Office of Research Oversight/Regulatory Affairs, Medical University of Innsbruck, Austria (EK Nr: 1064/2021). All participants provided written informed consent. Data were coded and anonymized, and all procedures complied with relevant ethical guidelines and the Declaration of Helsinki (https://www.wma.net/policies-post/wma-declaration-of-helsinki-ethicalprinciples-for-medical-research-involving-human-subjects/, accessed on 1 June 2020).
4.2. Whole Exome Sequencing (WES) and Variant Calling
Genomic DNA was isolated from buffy coat samples using standard protocols (Wizard Genomic DNA Purification Kit, Promega, Madison, WI, USA). PCR-free libraries were prepared from 1 µg DNA (TruSeq DNA PCR-Free HT Sample Preparation Kit, Illumina, San Diego, CA, USA) and sequenced at the NIH Intramural Sequencing Center (NISC, Bethesda, MD, USA) on the NovaSeq 6000 and NovaSeqX+ platforms (Illumina), generating ≥300 million paired-end reads (2 × 151 bp) per sample.
Sequence alignment and variant calling were performed using the Clara Parabricks Pipeline (v4.0.1). Reads were aligned to the hg38 reference genome with BWA MEM [32], duplicates were marked using Picard [33], and base quality score recalibration (BQSR) and variant calling were conducted with GATK HaplotypeCaller, generating gVCF files with a minimum Phred-scaled confidence threshold of 16.
4.3. SNV Detection from RNA-Seq Data
RNA-seq reads derived from buffy coat samples underwent quality assessment with FastQC (v0.11.9) and adapter/quality trimming using Trimmomatic (v0.36) [34]. Reads were aligned using a two-pass STAR workflow (v2.7.9a) [35] to hg19, followed by duplicate marking with Picard (v2.9.2) [33]. SNV calling was performed using the GATK RNA-seq variant discovery workflow, including base recalibration with known polymorphic sites (dbSNP138) and variant calling with GATK HaplotypeCaller in discovery mode (ERC gVCF, minimum Phred score 30).
Variants were filtered using standard hard-filtering criteria (QD < 2.0 || QUAL < 30.0 || SOR > 3.0 || FS > 60.0 || MQ < 40.0 || MQRankSum < −12.5 || ReadPosRankSum < −8.0) and further refined by excluding repetitive and blacklisted genomic regions [36,37]. Only heterozygous or homozygous alternate SNVs (0/1 or 1/1) with read depth ≥ 10, allele frequency ≥ 10%, and without excessive coverage were retained [38,39,40,41]. SNVs near indels (±5 bp) were excluded as likely artifacts. Genomic coordinates were lifted from hg19 to hg38.
4.4. RNA Sequencing (RNA-Seq) and Data Analysis
Total RNA (1 mg) was depleted of ribosomal RNA, reverse-transcribed, and used for library preparation with the TruSeq Stranded Total RNA Library Prep Kit with Ribo-Zero Gold (Illumina, RS-122-2301). Paired-end sequencing (2 × 150 bp) was performed on a NovaSeq 6000 system (Illumina).
After quality control (FastQC (v0.11.9) and Trimmomatic (v0.36) [34]), reads were aligned to hg38 using STAR (v2.7.9a) [35]. Gene-level counts were obtained with HTSeq (v0.9.1) [42], normalized, and analyzed for differential expression using DESeq2 [43,44]. Confounding technical variation was addressed using RUVSeq [45]. Genes with fewer than ten total reads were excluded. Statistical significance was assessed using a paired, two-sided Wilcoxon test with Benjamini–Hochberg correction. Genes with log_2_ fold change >1 or <−1, pAdj < 0.05, and without a 0 value in any sample were considered significant and subjected to gene enrichment analysis (GSEA). Enrichment significance was evaluated using the Kolmogorov–Smirnov statistic.
4.5. Allele Frequency Annotation and In Silico Functional Prediction
Population allele frequencies were obtained from dbSNP, gnomAD, All of Us, COSMIC, and ClinVar databases. The potential functional impact of JAK/STAT variants was assessed using AlphaMissense [15], Rare Exome Variant Ensemble Learner (REVEL) [17], and PolyPhen-2 [16], applying default scoring schemes for pathogenicity prediction.
4.6. Structural Analyses
The monomeric JAK3 protein structure was predicted using AlphaFold3 (https://alphafoldserver.com) and visualized and analyzed using PyMOL (v3.1.6.1).
5. Conclusions
This study addresses the framework of the context-dependent impact of rare genetic variants in immune pathways. It demonstrates that four very rare germline JAK/STAT variants (JAK3^P151R^, JAK3^R925S^, STAT5A^V494L^, and STAT6^Q633H^) can be inherited across generations without overt clinical manifestations, yet selectively associate with enhanced basal and virus-induced immune transcriptomes in a subset of carriers. Structural and in silico analyses suggest largely modest individual effects, while transcriptomic data support that immune activation emerges in a context-dependent manner, likely driven by combinatorial and epistatic interactions with additional immune regulatory variants such as TYK2. These findings highlight that JAK/STAT variants function as genetic modifiers rather than standalone drivers of immune phenotypes and underscore the necessity of family-based, genome-wide, and functional approaches to understand how multiple rare variants collectively shape human immune responses.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Hwa V. Human growth disorders associated with impaired GH action: Defects in STAT 5B and JAK 2Mol. Cell Endocrinol.202151911106310.1016/j.mce.2020.11106333122102 PMC 7736371 · doi ↗ · pubmed ↗
- 2Liu X. Robinson G.W. Wagner K.U. Garrett L. Wynshaw-Boris A. Hennighausen L. Stat 5a is mandatory for adult mammary gland development and lactogenesis Genes. Dev.19971117918610.1101/gad.11.2.1799009201 · doi ↗ · pubmed ↗
- 3Philips R.L. Wang Y. Cheon H. Kanno Y. Gadina M. Sartorelli V. Horvath C.M. Darnell J.E.Jr. Stark G.R. O’Shea J.J. The JAK-STAT pathway at 30: Much learned, much more to do Cell 20221853857387610.1016/j.cell.2022.09.02336240739 PMC 9815833 · doi ↗ · pubmed ↗
- 4Yao Z. Cui Y. Watford W.T. Bream J.H. Yamaoka K. Hissong B.D. Li D. Durum S.K. Jiang Q. Bhandoola A. Stat 5a/b are essential for normal lymphoid development and differentiation Proc. Natl. Acad. Sci. USA 20061031000100510.1073/pnas.050735010316418296 PMC 1327727 · doi ↗ · pubmed ↗
- 5Baxter E.J. Scott L.M. Campbell P.J. East C. Fourouclas N. Swanton S. Vassiliou G.S. Bench A.J. Boyd E.M. Curtin N. Acquired mutation of the tyrosine kinase JAK 2 in human myeloproliferative disorders Lancet 20053651054106110.1016/S 0140-6736(05)71142-915781101 · doi ↗ · pubmed ↗
- 6Levine R.L. Wadleigh M. Cools J. Ebert B.L. Wernig G. Huntly B.J. Boggon T.J. Wlodarska I. Clark J.J. Moore S. Activating mutation in the tyrosine kinase JAK 2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis Cancer Cell 2005738739710.1016/j.ccr.2005.03.02315837627 · doi ↗ · pubmed ↗
- 7Haapaniemi E.M. Kaustio M. Rajala H.L. van Adrichem A.J. Kainulainen L. Glumoff V. Doffinger R. Kuusanmaki H. Heiskanen-Kosma T. Trotta L. Autoimmunity, hypogammaglobulinemia, lymphoproliferation, and mycobacterial disease in patients with activating mutations in STAT 3Blood 201512563964810.1182/blood-2014-04-57010125349174 PMC 4304109 · doi ↗ · pubmed ↗
- 8Rajala H.L. Eldfors S. Kuusanmäki H. van Adrichem A.J. Olson T. Lagström S. Andersson E.I. Jerez A. Clemente M.J. Yan Y. Discovery of somatic STAT 5b mutations in large granular lymphocytic leukemia Blood 20131214541455010.1182/blood-2012-12-47457723596048 PMC 3668487 · doi ↗ · pubmed ↗