Genome-Wide Identification and Evolutionary Analysis of the bHLH Transcription Factor Family in Rosa roxburghii
Yuan-Yuan Li, Li-Zhen Ling, Shu-Dong Zhang

TL;DR
This study identifies and analyzes the bHLH transcription factor family in Rosa roxburghii, revealing evolutionary patterns and functional diversity.
Contribution
The study provides the first genome-wide analysis of bHLH transcription factors in Rosa roxburghii, highlighting lineage-specific evolution and functional divergence.
Findings
89 non-redundant RrbHLH genes were identified and classified into 23 subfamilies.
Segmental duplication and structural variations contributed to functional divergence in RrbHLH genes.
Promoter analysis revealed cis-acting elements linked to stress and hormone responses.
Abstract
The basic/helix-loop-helix (bHLH) transcription factors are crucial regulators of plant development and stress responses. In this study, we conducted a genome-wide analysis of the bHLH family in Rosa roxburghii, an economically important fruit crop. A total of 89 non-redundant RrbHLHs were identified and unevenly distributed across the seven chromosomes. Phylogenetic analysis classified them into 23 subfamilies and 7 Arabidopsis subfamilies were absent, indicating lineage-specific evolutionary trajectories. Conserved motif and gene structure analyses showed that members within the same subfamily generally shared similar architectures, yet subfamily-specific variations were evident, suggesting potential functional diversification. Notably, key residues involved in DNA-binding and dimerization were highly conserved within the bHLH domain. Promoter analysis identified multiple cis-acting…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
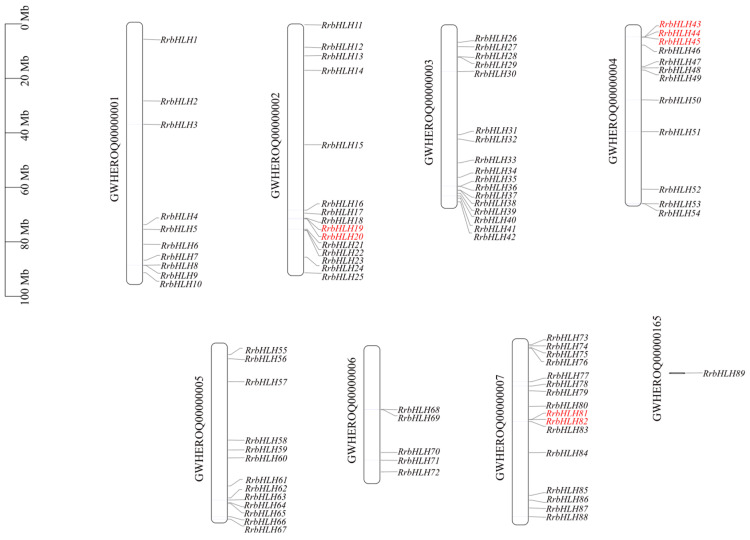 Figure 1
Figure 1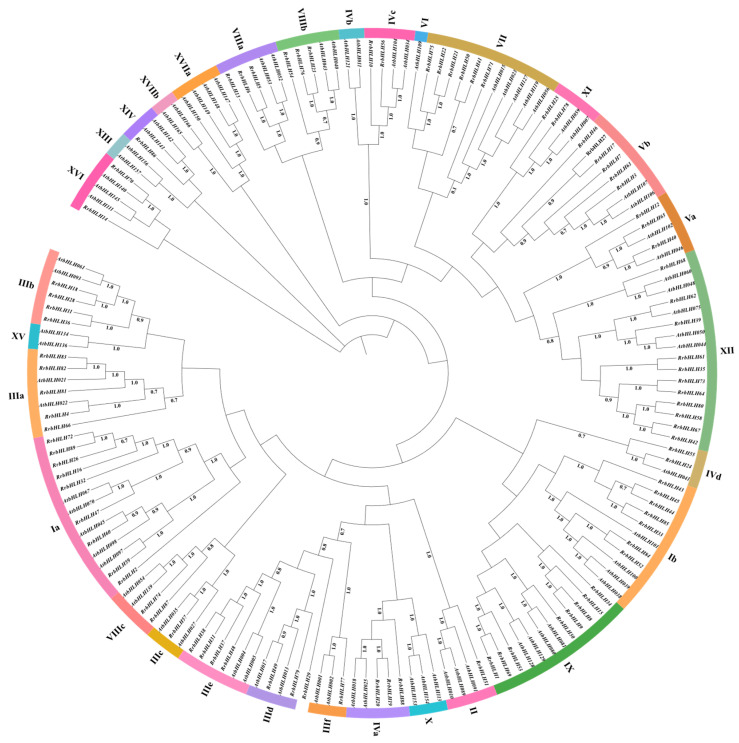 Figure 2
Figure 2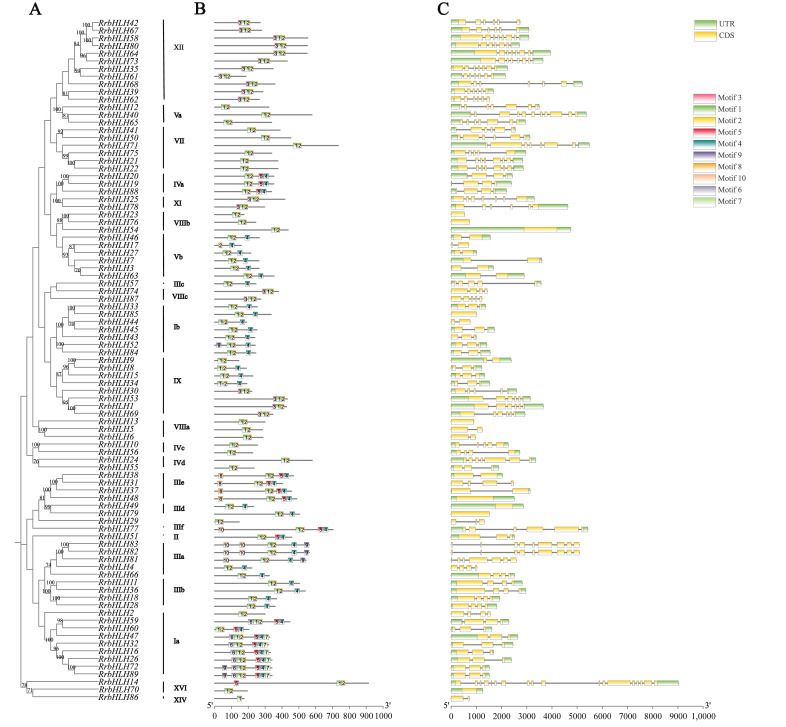 Figure 3
Figure 3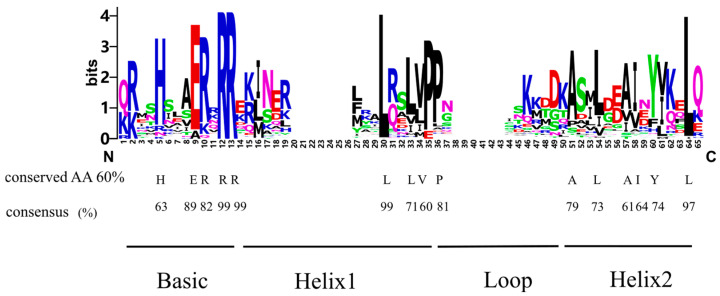 Figure 4
Figure 4 Figure 5
Figure 5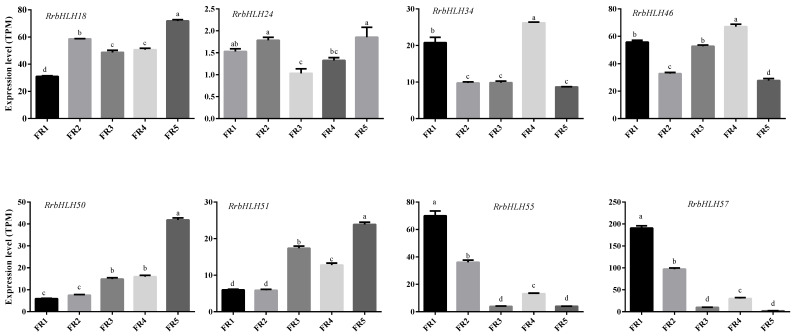 Figure 6
Figure 6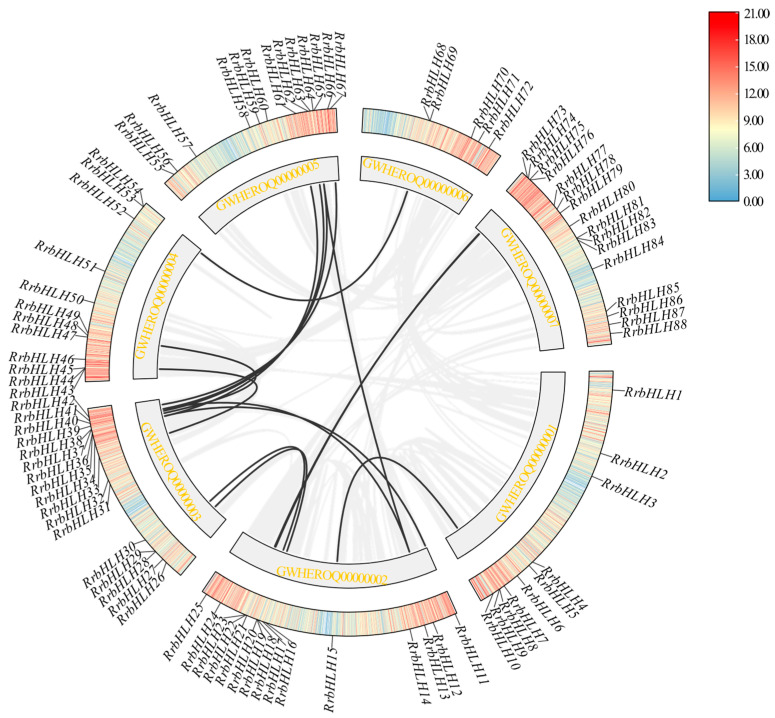 Figure 7
Figure 7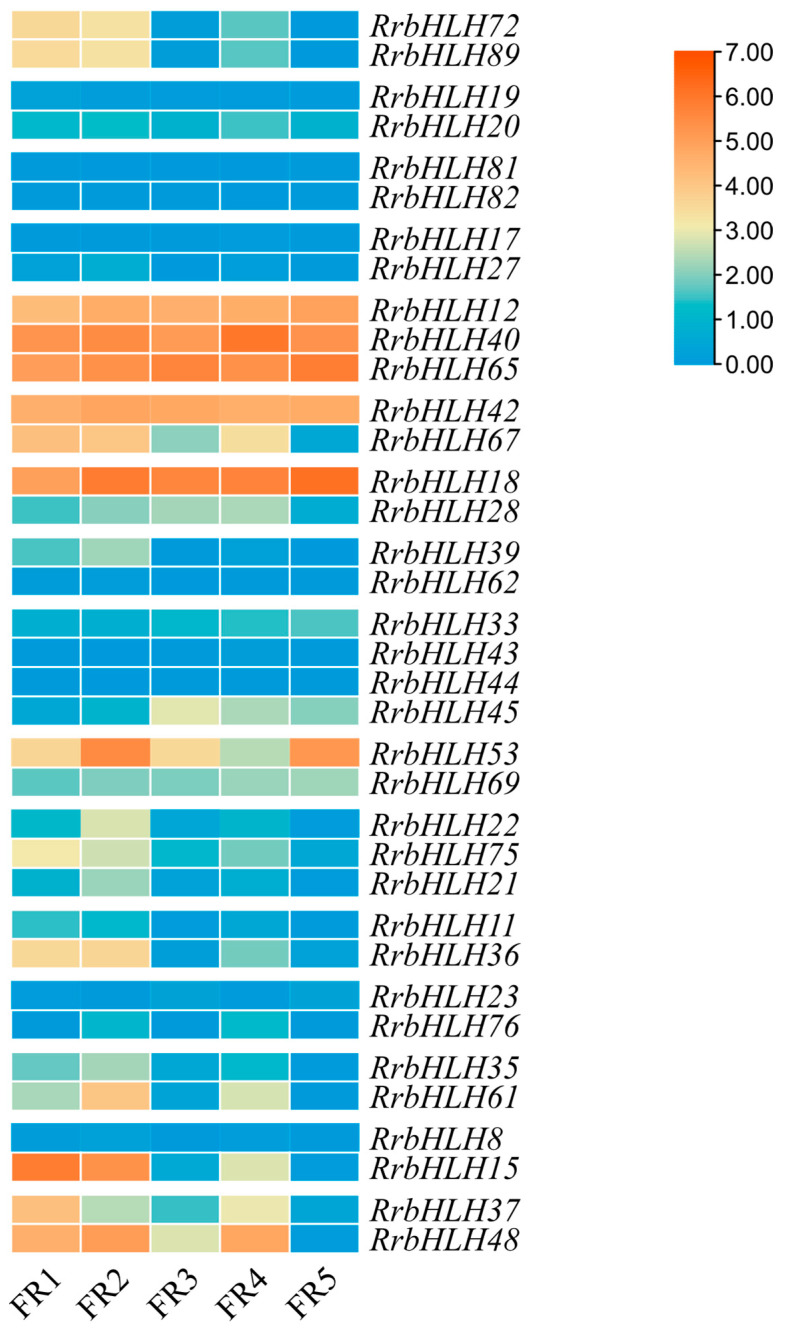 Figure 8
Figure 8 Figure 9
Figure 9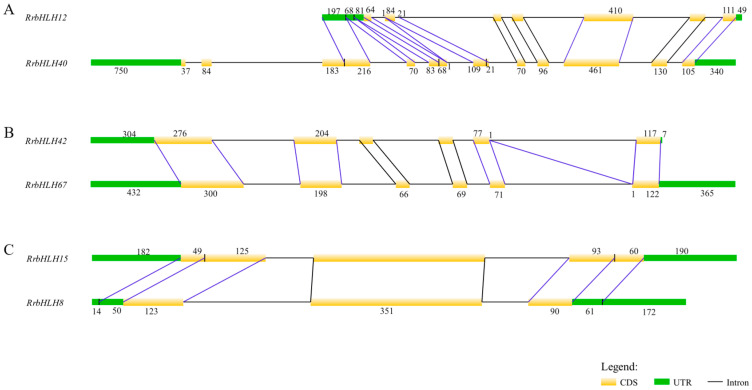 Figure 10
Figure 10- —Science and Technology Program of Liupanshui
- —Liupanshui Normal University Postgraduate Special Project
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPlant Gene Expression Analysis · Plant Molecular Biology Research · Photosynthetic Processes and Mechanisms
1. Introduction
The basic/helix-loop-helix (bHLH) transcription factors represent a substantial and evolutionarily conserved superfamily of regulatory proteins in eukaryotes [1,2]. A typical bHLH domain encompasses approximately 60 amino acids and comprises two functionally distinct regions: an N-terminal basic region that recognizes and binds to specific DNA motifs, such as the E-box (CANNTG) in target gene promoters, and a C-terminal helix-loop-helix (HLH) region that facilitates protein dimerization [3,4,5]. As the second-largest transcription factor family in plants, bHLHs are crucial in various biological processes, including growth and development, secondary metabolism, hormone signaling, and responses to biotic and abiotic stresses [1,6]. For instance, bHLH members in Arabidopsis thaliana such as FIT and PYE are key regulators of iron homeostasis, while MYC2 and TT8 are involved in jasmonic acid signaling and anthocyanin biosynthesis, respectively [7].
The functional diversity of the bHLH family is intricately linked to its structural complexity and evolutionary history, particularly through gene duplication events. Whole-genome duplication (WGD), segmental duplication, and tandem duplication have been identified as significant drivers of the expansion of the bHLH family across plant species [8]. For example, studies on pummelo (Citrus grandis) [9] and wintersweet (Chimonanthus praecox) [10] have revealed that 47 and 56 bHLH genes, respectively, originated from segmental duplication events. These duplication events provide a genetic reservoir for functional diversification, where some genes retain ancestral functions while others undergo neofunctionalization or subfunctionalization due to sequence variation and divergence in expression patterns. Furthermore, bHLH proteins often interact with other transcription factors, such as MYB and WD40 proteins, to form multi-protein complexes (e.g., the MBW complex) that coordinately regulate the biosynthesis of secondary metabolites, including anthocyanins and floral volatiles [11]. This interaction enhances their regulatory potential in plant adaptation and trait development [12,13].
Rosa roxburghii Tratt., commonly known as Cili or chestnut rose, is a perennial shrub of the Rosaceae family valued for its nutritional and medicinal fruit. The fruit is particularly rich in vitamin C and often referred to as the ‘King of Vitamin C’, and also contains other bioactive compounds like polyphenols and flavonoids [14,15]. Recent advancements, notably the release of the R. roxburghii genome sequence [16], have accelerated research into the molecular mechanisms underlying its key agronomic traits, including L-ascorbic acid content, fruit development, stress tolerance, and the accumulation of secondary metabolites [17,18,19,20]. Comparative analysis of bHLH genes across Rosaceae species indicate that the expansion of the bHLH gene family in this lineage has been driven by both ancient and recent duplication events. Furthermore, these genes have been shown to play significant roles in responses to abiotic stresses such as cold and drought [21,22,23]. Despite these advancements, a systematic investigation of the bHLH transcription factor family in R. roxburghii remains limited, resulting in a knowledge gap in our understanding of the genetic regulation underlying its economically important traits.
To address this research gap, we conducted a systematic genome-wide analysis of the bHLH transcription factor family in R. roxburghii. The specific objectives of this study were as follows: (1) identify and characterize all putative bHLH genes along with their physicochemical properties; (2) examine their phylogenetic relationships, gene structures, conserved motifs, and chromosomal distributions; (3) investigate gene duplication events and evolutionary constraints; (4) analyze their expression profiles in developing fruits. Our results provide the first comprehensive genomic characterization of the RrbHLH gene family, offering insights into the evolutionary dynamics underlying its functional diversification. This work establishes a solid foundation for future functional genomic studies and facilitates molecular breeding strategies for the genetic improvement of this economically important fruit crop.
2. Results
2.1. Identification and Physicochemical Characterization of the RrbHLH Gene Family
A genome-wide identification of bHLH members in R. roxburghii was conducted using integrated BLAST and HMMER searches using TBtools (v2.371), followed by validation of the conserved bHLH domain via the Pfam and SMART database. This comprehensive analysis identified 89 non-redundant bHLH transcription factors within the R. roxburghii genome. Chromosomal mapping revealed that 88 RrbHLH genes were distributed across chromosomes 1 to 7, with gene counts per chromosome ranging from 5 to 17 (Figure 1; Table S1). These genes were systematically designated as RrbHLH1 to RrbHLH88 based on their chromosomal positions, while the remaining gene (RrbHLH89) was located on a genomic scaffold.
Subsequent analyses of the physicochemical properties of RrbHLH proteins revealed substantial variation among family members. The lengths of these proteins ranged from 142 amino acids (RrbHLH9) to 912 amino acids (RrbHLH14), with corresponding molecular weights varying from approximately 16.39 kDa to 100.19 kDa (Table S1). Theoretical isoelectric points (pI) ranged from 4.71 to 10.32, indicating diverse charge characteristics at physiological pH (Table S1). Analysis of the instability index suggested that the majority of RrbHLHs exhibited values exceeding 40, thus classifying them as unstable. Predictions of subcellular localization indicated that 83 RrbHLHs are localizated in the nucleus, consistent with their anticipated function as transcription factors. The remaining six proteins demonstrated alternative localizations: two each in cytosol–nucleus dual localization (RrbHLH9, RrbHLH60), cytosol-only (RrbHLH34, RrbHLH44), and chloroplast (RrbHLH17, RrbHLH35). Collectively, these findings reveal considerable diversity in the physicochemical properties of the RrbHLH family members.
2.2. Phylogenetic Analysis of RrbHLHs
To elucidate the evolutionary relationships among RrbHLH genes, we constructed a phylogenetic tree utilizing full-length protein sequences from 89 R. roxburghii and 76 AtbHLH genes. The bHLH genes were classified into 17 families, comprising a total of 30 subfamilies (Figure 2). These RrbHLH members were distributed across 23 subfamilies, while 7 subfamilies (IVb, VI, X, XIII, XV, XVIIa, and XVIIb) lacked representatives of RrbHLH genes. Subfamily XII contained the highest number of RrbHLH members, followed by subfamilies Ia and IX (Figure 2 and Figure 3A). In contrast, subfamilies II, IIIc, IIIf, and XIV each contained only a single RrbHLH member. Additionally, RrbHLH29 was not assigned to any established subfamily and was therefore designated as an orphan gene (Figure 2 and Figure 3A).
To explore the potential functional diversification of RrbHLHs from an evolutionary perspective, we mapped the distribution of functionally annotated bHLH orthologs (defined by NCBI CDD, e.g., bHLH_AtPIF_like, bHLH_AtAMS_like) onto our phylogenetic subfamilies (Figure S1). This analysis revealed distinct evolutionary patterns linked to functional specialization. RrbHLH orthologs of functionally niche types, such as bHLH_AtPIF_like, were exclusively clustered within specific subfamilies (e.g., Subfamily VII), suggesting their evolutionary trajectory towards specialized environmental sensing, like light perception. Conversely, orthologs of proteins with bHLH_AtbHLH_like were found in distinct lineages (Subfamilies VIIIa and IX). Furthermore, functions related to stress response and specialized metabolism (e.g., bHLH_AtAIB_like, bHLH_AtMYC1_like) primarily arose through internal diversification within large, ancient subfamilies. Most notably, Family III harbors diverse roles ranging from hormone signaling (Subfamily IIId) to anthocyanin biosynthesis (Subfamily IIIf). Collectively, the functional landscape of RrbHLH proteins appears to be shaped by three core evolutionary strategies: lineage specialization, convergent recruitment, and internal radiation.
2.3. Motif Composition and Gene Structure of the RrbHLH Family
The conserved motif analysis of the 89 RrbHLH protein sequences revealed a total of 10 distinct motifs, designated as motif1 through motif10. The detailed information of these motifs was presented in Figure S2. Notably, these motifs with varying compositions among the different protein members (Figure 3B). Motif1 and motif2, which form the core bHLH domain, were found adjacent to each other in the sequence. Six subfamilies—Va, VII, VIIIa, VIIIb, IVc, and IVd—contained only these two motifs, with the exceptions of RrbHLH17, which possesses only motif2, and RrbHLH86, which possesses only motif1. Most subfamilies exhibited characteristic motif patterns; for example, all 11 members of subfamily XII contained motifs 1, 2, and 3, while subfamily IIIb contained motifs 1, 2, and 4 (Leucine Zipper). Variations were also noted within certain subfamilies: although most members of subfamily Ib contained motifs 1, 2, and 4, RrbHLH52 additionally included motif 9 (Zinc Finger). A similar pattern was observed in subfamily Vb. Furthermore, in subfamily IX, some members contained motifs 1, 2, and 4, while others contained motifs 1, 2, and 3.
Gene structure analysis revealed that the lengths of RrbHLH genes ranged from 522 bp to 9024 bp, with varying numbers of exons (Table S1). RrbHLH14 exhibited the highest number of exons (18), while nine genes were found to be intronless (Figure 3C). Most members within the same subfamily displayed similar exon–intron organizations. For example, all members of subfamilies Vb, VIIIb, and IVa consistently contained 2, 1, and 4 exons, respectively. Additionally, most members of subfamilies Ia and Ib shared similar structural patterns. In contrast, members of other subfamilies showed considerable structural divergence (Figure 3C). Collectively, these results demonstrate the complex gene structure of the RrbHLH family.
2.4. Characterization of Key Amino Acid Residues in Conserved bHLH Region
To predict DNA-binding properties, we aligned the amino acid sequences of the bHLH domain of RrbHLH proteins. The results show that the first 13 amino acids correspond to the basic region. Among RrbHLH proteins, 89% contain a glutamic acid (E) residue at position 9, which directly interacts with the CA nucleotides of the hexanucleotide E-box sequence (Figure 4). Furthermore, more than 60% of the proteins exhibit a histidine (H) at position 5 and an arginine (R) at position 13, forming the characteristic configuration H5-E9-R13 that interacts with the canonical hexanucleotide E-box motif (5′-CANNTG-3′). Other frequently occurring basic residues, such as arginine at positions 10 and 12, are also highly conserved in RrbHLH proteins, with consensus rates of 82% and 99%, respectively (Figure 4).
The HLH region facilitates the formation of homodimeric or heterodimeric complexes between bHLH proteins. Our analysis indicated that the hydrophobic residues Leu-30 in helix 1 and Leu-64 in helix 2 are the most conserved across the RrbHLH family (99% and 97%, respectively) (Figure 4). Additionally, a conserved proline (P) interrupts the first helix and initiates a loop of variable length, typically consisting of six to eight residues.
2.5. Cis-Acting Element Analysis in Promoter of RrbHLHs
To investigate the transcriptional regulation of RrbHLH genes, we analyzed the 2000 bp upstream promoter sequences of all 89 bHLH members, identifying a total of 2435 cis-acting elements. Each RrbHLH gene exhibited multiple types of cis-acting elements, with light-responsive elements found in all members, indicating a conserved potential for light-mediated regulation across the family (Figure 5). These elements were subsequently classified into five functional categories.
Hormone-responsive elements, including ABRE, TCA-element, TGACG-motif, and CGTCA-motif, are associated with responses to the abscisic acid, salicylic acid, methyl jasmonate, and other phytohormones, suggesting their roles in hormonal signaling during growth and development. Stress-responsive elements such as LTR, MBS, and TC-rich repeats are implicated in adaptation to low temperature, drought, and defense responses. Tissue-specific elements, including GCN4_motif and HD-Zip 1, may regulate expression in specific organs or cell types, such as endosperm, root, meristem, and mesophyll tissues. Metabolism-related elements, including MBSI and O2-site, are potentially involved in the regulation of flavonoid biosynthesis and zein metabolism regulation. Additionally, core promoter elements such as the CAAT-box and enhancer regions, along with transcription factor-binding sites like those for MYB, were identified as fundamental components of the transcriptional machinery. This systematic profiling reveals a complex regulatory architecture of RrbHLH genes and provides insights into their potential functions in plant development, stress adaptation, and metabolic regulation.
2.6. Expression Profiling of the RrbHLH Gene Family in Rosa roxburghii
To further understand the functional diversity of RrbHLH genes, we analyzed their expression patterns during fruit development. Initially, we compared the expression levels between GA_3_-treated and untreated fruits at 120 days post-pollination. Hierarchical clustering analysis categorized the 89 RrbHLH genes into three distinct clusters based on their expression profiles (Figure S3). Approximately half of the RrbHLH genes (41 genes) exhibited low expression levels, including RrbHLH16, RrbHLH60, RrbHLH19, and RrbHLH85. In contrast, 23 genes, such as RrbHLH42 and RrbHLH78, demonstrated consistently high expression. The remaining genes displayed moderate expression levels.
A total of 30 RrbHLH genes exhibited significant differential expression in fruits following GA_3_ treatment (Figure S3). These genes were categorized into 16 subfamilies, including four from subfamily XII, and three each from subfamilies Ia, IIIb, IIIe, and IX (Figure 1 and Figure S3). Notably, four RrbHLH genes from different subfamilies—RrbHLH57 (IIIc), RrbHLH24 (IVd), RrbHLH55 (IVd), and RrbHLH34 (IX)—demonstrated increased expression levels.
We further examined the expression patterns of RrbHLH genes across various stages of fruit development. A total of 75 RrbHLH genes exhibited dynamic expression changes during this process, which allowed us to categorize them into two distinct clusters (Figure S4). Notably, we concentrated on genes that showed significant expression differences following GA_3_ treatment (Figure S3). For instance, RrbHLH55 and RrbHLH57 were relatively highly expressed during the early developmental stage FR1, while RrbHLH34 and RrbHLH46 showed elevated expression levels in FR4. By contrast, RrbHLH18, RrbHLH24, RrbHLH50, and RrbHLH51 showed higher expression in FR5 (Figure 6). These results suggest that these genes may have distinct roles at different stages of fruit development.
2.7. Gene Duplication Events and Functional Divergence of RrbHLHs
To elucidate the expansion mechanism of the RrbHLH gene family, we analyzed gene duplication events using MCScanX. A total of 36 RrbHLH genes were identified as products of duplication events, forming 21 duplicated pairs. They included 4 tandem duplication pairs primarily located on chromosomes 2, 4, and 7 (Figure 1), as well as 17 segmental duplication pairs involving 29 genes distributed across all seven chromosomes (Figure 7). All duplicated pairs clustered within the same subfamily (Table S2; Figure 2); for instance, RrbHLH19/20 belongs to subfamily IVa, while RrbHLH81/82 is part of subfamily IIIa. Notably, RrbHLH44 formed separately duplication pairs with RrbHLH43 and RrbHLH45. One segmental pair, RrbHLH72/89, included a gene located on a scaffold (Figure 1). These findings suggest that segmental duplication served as the primary driver of expansion in the RrbHLH family. Selection pressure analysis revealed that all calculable duplicated pairs exhibited non-synonymous (Ka) and synonymous (Ks) substitutions rates below 1, indicating the action of purifying selection during evolution (Table S2). The Ka/Ks ratio for the segmental pair RrbHLH37/48 could not be determined due to high sequence divergence. Divergence times of the duplicated pairs, estimated from Ks values, ranged from approximately 41.07 to 121.23 million years ago.
We analyzed the expression patterns of duplicated gene pairs across various stages of fruit development. The results showed that the two tandem duplication pairs (RrbHLH43/RrbHLH44 and RrbHLH81/RrbHLH82) exhibited no detectable expression. In contrast, one segmental duplication pair (RrbHLH72/RrbHLH89) displayed similar expression profiles (Figure 8). However, most segmental duplication pairs demonstrated divergent expression patterns during fruit development. For example, RrbHLH42 maintained relatively high expression across all stages, while its duplicated counterpart RrbHLH67 exhibited a decreasing trend during the first three stages, followed by an increase at the fourth stage and a subsequent decline at the fifth stage. These findings suggest that duplications have significantly contributed to the functional diversification within the RrbHLH family.
2.8. Co-Expression Networks of Duplicated RrbHLH Pairs
To further investigate the expression regulatory patterns of duplicated RrbHLH pairs, we constructed a weighted gene co-expression network (WGCNA). A total of 22,544 genes were grouped into 33 distinct modules with 58 RrbHLHs distributed across 16 modules (Figure S5). Among these, 22 duplicated RrbHLHs were allocated to 13 modules, with most gene pairs assigned to different modules (Table S2), suggesting divergence in their biological functions. For instance, RrbHLH18 and RrbHLH28 were located in the midnight blue and light yellow modules, respectively (Table S2). Notably, this duplicated pair also showed differential expression levels across various fruit developmental stages (Figure 8).
We analyzed the correlation between the 33 modules and eight tissue types (Figure 9A). The results revealed that duplicated RrbHLHs present in the 13 modules were correlated with all tissues except FR3. For example, RrbHLH72 and RrbHLH89 were clustered in the red module and were primarily associated with flower bud (Table S2 and Figure 9A). Meanwhile, RrbHLH11 and RrbHLH36 were assigned to the green and purple modules, respectively, and both separately showed correlation with FR4 and leaf (Table S2 and Figure 9B,C). Analysis of co-expressed genes further indicated that the RrbHLH11/36 duplicated pair participates in distinct gene co-expression networks (Figure 9B,C). Together, these findings suggest that gene duplication has contributed to functional divergence within the RrbHLH family.
2.9. Exon–Intron Structural Divergence Among Duplicated RrbHLH Pairs
Structural analysis revealed a significant divergence in the exon–intron organization among the 21 duplicated RrbHLH gene pairs. Of these, 7 pairs (33.3%) exhibited differences in exon numbers, indicative of major structural reorganization, while the remaining 14 pairs (66.7%) displayed length variations in one or more homologous exons despite maintaining identical exon counts (Figure S6). Further investigation through precise sequence alignment identified three fundamental mechanisms driving these structural variations. First, intraexonic indels were universally observed across all duplicated pairs, manifesting as small insertions or deletions within exons that directly contribute to exon length divergence; complete exon indels were particularly rare and detected only in pairs involving RrbHLH40/12, RrbHLH40/65, and RrbHLH81/82 (Figure 10). Second, the widespread phenomenon of exon dissolution and joining occurred in 20 of the 21 pairs, primarily involving either the splitting of single exons into multiple smaller exons or the merging of multiple exons into larger units. An example of this is the alignment of the fifth exon of RrbHLH42 with both the fifth and sixth exons of RrbHLH67 (Figure 10). Third, the processes of exonization and pseudoexonization facilitated the interconversion between exonic and non-exonic sequences, with a particularly illustrative example being the alignment of terminal exon regions in RrbHLH15 with untranslated regions in RrbHLH8 (Figure 10). Collectively, these structural divergences, particularly those affecting exon composition and length, establish a plausible molecular basis for the functional differentiation observed among duplicated RrbHLH gene pairs.
3. Discussion
The bHLH transcription factor family is one of the largest and most functionally diverse gene families in plants, playing pivotal roles in regulating growth, development, stress responses, and secondary metabolism [8,24]. Recent advancements in high-throughput sequencing have enabled the genome-wide characterization of gene families in non-model species. Utilizing the recently released genome of R. roxburghii, this study identified 89 RrbHLHs in this nutritionally valuable fruit crop.
Sequence analysis of RrbHLH proteins confirmed a high degree of conservation at functionally critical positions within the bHLH domain. Key residues in the basic region—including H5, E9, and R13, which are involved in direct DNA contact—were broadly conserved, as were hydrophobic leucine residues in the HLH region that stabilize the dimerization interface [24,25,26]. This conservation indicates that the core DNA-binding and dimerization functions of the ancestral bHLH domain have been maintained in R. roxburghii. Beyond this conserved core, several RrbHLH members contain additional functional motifs, such as leucine zippers, which may fine-tune dimerization specificity, transcriptional activity, or protein stability. Phylogenetic analysis revealed that these accessory domains are restricted to particular subfamilies (e.g., Ia, IIIa and IIIe), consistent with independent acquisition events during evolution. This pattern supports the occurrence of domain shuffling in the expansion of the bHLH family, as reported in broader studies of plant bHLH proteins [24,26]. The modular addition of such domains likely provided a structural foundation for functional diversification, allowing RrbHLH proteins to participate in more specialized regulatory networks during plant development and stress responses.
The bHLH transcription factor family has undergone significant expansion during plant evolution [24]. In R. roxburghii, we identified 89 bHLH genes, a number comparable to some diploid Rosaceae species but smaller than in Arabidopsis [7] or rice [22], which may be attributed to variations in genome size, ploidy, and lineage-specific duplication events. Gene duplication has been a primary driver the expansion of the bHLH family [27,28]. In R. roxburghii, we detected 21 duplicated gene pairs, comprising 17 segmental and 4 tandem duplication pairs. This pattern aligns with the expansion mechanisms observed in other Rosaceae species [23]. Comparative analysis across five Rosaceae species revealed that WGD and segmental duplication played critical roles in pear and apple, whereas dispersed duplication was more prominent in peach, strawberry, and Chinese plum [23]. The estimated divergence times of the RrbHLH duplication pairs, based on Ks values, span a wide range (41121 MYA), reflecting multiple rounds of duplication throughout evolutionary history. Our findings align with broader trends observed in Rosaceae bHLH research. For example, studies in pear identified 198 PbbHLH genes, with expansion driven largely by recent WGD events (3045 MYA), while peach, strawberry, and Chinese plum experienced more ancient duplication events [23]. Notably, seven Arabidopsis subfamilies (IVb, VI, X, XIII, XV, XVIIa, XVIIb) are absent in R. roxburghii, possibly due to lineage-specific gene loss or functional consolidation.
After gene duplication, copies can acquire novel functions or undergo division of labor through sequence variation, expression divergence, and structural reorganization. In this study, most duplicated RrbHLH pairs exhibited Ka/Ks ratios of less than 1, indicating the action of purifying selection. Similar patterns were observed in the bHLH families of the Rosaceae, where most gene pairs evolved under negative selection [23]. Several RrbHLH segmental pairs displayed distinct expression profiles across various fruit developmental stages, implying subfunctionalization or neofunctionalization. Structural analysis further revealed three main types of post-duplication variation: (i) intraexonic insertions/deletions, (ii) exon dissolution/joining, and (iii) exonization/pseudoexonization. These structural alterations could influence mRNA splicing, stability, or protein domain architecture [29], thereby providing a molecular basis for functional differentiation between duplicates.
Members of the same bHLH subfamily are frequently involved in analogous biological processes. Most bHLH proteins in Arabidopsis have been functionally characterized [7,30] and the functions of RrbHLHs will be discussed herein in the context of the corresponding genes from Arabidopsis. The best-described bHLH proteins are members of subgroup IIIf, which were involved in the MBW complexes to regulate flavonoid biosynthesis, the differentiation of trichomes, root hair cells and the biosynthesis of seed coat mucilage [7] (Table S3). Therefore, RrbHLH77 from subfamily IIIf may also be involved in these functions, akin to those observed in Arabidopsis (Table S3). A striking exception comes from three A. thaliana subfamily Ia proteins, AtbHLH045, AtbHLH097 and AtbHLH098, which play nonoverlapping roles in controlling sequential cell fate specification during stomatal differentiation (Table S3). Such apparent functional redundancy is also observed in subfamilies Ib and II, which were involved in iron homeostasis and anther development, respectively. Furthermore, an outstanding example of functional diversification is encountered in subfamily XII, which clusters five AtbHLHs and eleven RrbHLHs involved in diverse biological roles, including brassinosteroid signaling, phytochrome-dependent photomorphogenic responses (shade avoidance), development of the female reproductive tract, responses to salinity and drought stress, cold stress response and hypocotyl elongation (Table S3). Therefore, future research should prioritize the functional characterization of key RrbHLH members, particularly those associated with fruit development, stress adaptation, or metabolite biosynthesis, utilizing genetic and molecular approaches.
4. Materials and Methods
4.1. Identification of the bHLH Gene Family in Rosa roxburghii
The genome sequence and corresponding annotation files of R. roxburghii were retrieved from the China National Center for Bioinformation (https://www.cncb.ac.cn/; accession GWHEROQ00000000). Reference protein sequences of bHLH transcription factors from Arabidopsis thaliana and Oryza sativa were obtained from the PlantTFDB database (https://planttfdb.gao-lab.org/, accessed on 10 July 2025) to serve as queries. A local BLASTP search was conducted against the complete R. roxburghii proteome using TBtools (v2.371) with the bHLH protein sequences from A. thaliana and O. sativa separately, applying an E-value cutoff of 1 × 10^−5^. The resulting hits from both searches were merged and deduplicated to generate a preliminary candidate set. Concurrently, the Hidden Markov Model (HMM) profile of the bHLH domain (PF00010) was acquired from the Pfam database and used to scan the R. roxburghii proteome via the embedded HMMER tool in TBtools (v2.371) [31,32]. Candidate genes identified through both BLAST and HMMER approaches were consolidated into a non-redundant set. The corresponding protein sequences were extracted and further validated using the Pfam (https://www.ebi.ac.uk/interpro/, accessed on 12 July 2025), SMART and NCBI Conserved Domains Database (CDD) to verify the presence of a complete bHLH domain. Any sequences lacking a confirmed bHLH domain were excluded, yielding a final set of high-confidence RrbHLH genes.
4.2. Analysis of Physicochemical Properties, Conserved Motifs, and Gene Structures
The physicochemical properties of the identified RrbHLH proteins, including the number of amino acids (aa), isoelectric point (pI), and molecular weight (MW), were computed using the ‘Protein Parameter Calc’ module in TBtools (v2.371). Conserved motifs within the RrbHLH protein sequences were predicted using the ‘Simple MEME Wrapper’ function in TBtools, with the parameters set to a maximum of 10 motifs and an optimum motif width ranging from 6 to 50 amino acids, while all other settings remained at their default values. Gene structure features, specifically the exon–intron organization of each RrbHLH gene, were extracted from the R. roxburghii genome annotation file in GFF3 format. Visualization of conserved motif distributions and gene structures was subsequently conducted using the ‘Visualize MEME Motif Pattern’ and ‘Visualize Gene Structure’ tools in TBtools, respectively.
4.3. Promoter Cis-Acting Element Analysis
The 2000 bp genomic sequences located upstream of the transcription start site of each RrbHLH gene were retrieved as putative promoter regions using TBtools (v2.371). These promoter sequences were screened for cis-acting elements through the PlantCARE database (https://bioinformatics.psb.ugent.be/webtools/plantcare/html/, accessed on 20 July 2025). The identified cis-acting elements were subsequently categorized, and their distributions were visualized using TBtools (v2.371).
4.4. Phylogenetic Analysis and Chromosomal Distribution of RrbHLHs
A total of 76 representative AtbHLH protein sequences (see Table S3) were retrieved from the TAIR database, covering all known subfamilies [7]. Multiple sequence alignment of the RrbHLH proteins and AtbHLH representatives was performed using the ClustalW algorithm in MEGA software (v12) with default parameters [33]. A neighbor-joining (NJ) phylogenetic tree was constructed from the aligned sequences using pairwise deletion, the p-distance method, and 1000 bootstrap replicates in MEGA (v12) [33]. The resulting tree was visualized and annotated using the iTOL online platform (https://itol.embl.de/, accessed on 2 August 2025), with the Newick format output from MEGA serving as the input. For chromosomal distribution analysis, the physical locations of the RrbHLH genes were mapped onto chromosomes using TBtools (v2.371), based on positional information extracted from the R. roxburghii genome annotation file in GFF format.
4.5. Gene Duplication and Selection Pressure Analysis
Intra-genomic synteny analysis was conducted to identify duplication events within the RrbHLH gene family using the MCScanX algorithm implemented in TBtools (v2.371). Both segmental and tandem duplications were detected, and the syntenic relationships among RrbHLH genes were visualized using a Circos plot [34]. For each duplicated gene pair, the rates of non-synonymous (Ka) and synonymous (Ks) substitutions were calculated using the ‘Simple Ka/Ks Calculator’ in TBtools. The Ka/Ks ratio was used to assess selective pressure, with interpretations as follows: Ka/Ks < 1 indicates purifying selection, Ka/Ks ≈ 1 suggests neutral evolution, and Ka/Ks > 1 reflects positive selection. The divergence time (T) for each duplicated pair was estimated using the equation T = Ks/(2 × r), where a substitution rate (r) of 1.5 × 10^−8^ substitutions per site per year was applied for dicot plants.
4.6. Expression Profiling Based on Transcriptomic Data
RNA-seq data for R. roxburghii were obtained from two publicly available datasets. The first dataset, retrieved from the NCBI SRA database (accession PRJNA1003688), comprised transcriptomes of GA_3_-treated and untreated fruits collected 120 days post-pollination, each with three biological replicates. The second dataset, acquired from the Genome Sequence Archive at the National Genomics Data Center (accession CRA017453), included samples collected across different stages of fruit development. Following quality control, clean reads were aligned to the R. roxburghii reference genome using HISAT2 [35]. The expression levels of RrbHLH genes were quantified and normalized as transcripts per million (TPM). A heatmap was subsequently generated using TBtools (v2.371) to visualize expression patterns across tissues, and hierarchical clustering was employed to group genes with similar expression profiles.
4.7. Weighted Correlation Network Analysis (WGCNA)
The top 50% of transcripts ranked by the expression levels were selected for WGCNA. In this study, we conducted the analysis from 9 samples of leaf, flower and flower bud, except for the abovemetioned 15 different developing fruit tissues. The pickSoftThreshold function was applied to determine an appropriate soft-thresholding power, ensuring that the resulting network followed a scale-free topology. Based on pairwise gene correlations raised to this power, an adjacency matrix was constructed and subsequently converted into a topological overlap matrix (TOM). Module detection was carried out using the dynamic tree cut method, with the following parameters: minModuleSize = 30, mergeCutHeight = 0.15, and deepSplit = 2. The network was configured as “unsigned.”
4.8. Structural Divergence Analysis of Duplicated Gene Pairs
Structural divergence among paralogous genes was assessed by comparing their exon-intron organizations in accordance with established methodologies [36]. Gene pairs were classified as structurally divergent if they displayed either differing exon counts or identical exon counts with divergent lengths in at least one homologous exon. To elucidate the mechanisms driving these structural variations, pairwise alignments of genomic sequences were performed using the corresponding cDNA sequences as reference. Through meticulous sequence comparison, three distinct types of structural divergence events were identified: (1) Intraexonic indels, characterized by insertions or deletions within homologous exons; (2) Exon dissolution/joining events, where one exon is split into multiple smaller exons or multiple exons are merged into a single larger exon; and (3) Exonization/pseudoexonization, which involves the evolutionary transition between exonic and intronic sequences among paralogs.
5. Conclusions
In this study, we conducted a comprehensive genome-wide analysis of the bHLH transcription factor family in R roxburghii, leading to the identification and characterization of 89 non-redundant RrbHLH genes. Through phylogenetic analysis with Arabidopsis thaliana, these members were classified into 23 subfamilies, with 7 subfamilies (IVb, VI, X, XIII, XV, XVIIa, and XVIIb) notably absent, suggesting lineage-specific evolutionary trajectories. The bHLH members within the same subfamily generally shared similar exon-intron architectures and motif compositions. Subfamily-specific and intra-subfamily variations were observed, highlighting the structural basis for functional diversification. Key DNA-binding and dimerization residues in the bHLH domain were highly conserved, preserving the core molecular functions of this domain. Expression profiling revealed dynamic and diverse expression patterns across fruit developmental stages and in response to GA_3_ treatment. Gene duplication analysis identified 21 duplicated pairs, primarily driven by segmental duplication. Although all pairs evolved under purifying selection, their divergent expression patterns and structural variations—including exon indels, dissolution/joining, and exonization/pseudoexonization—provide a mechanistic basis for functional divergence. Collectively, these findings offer fundamental insights into the evolution and potential functions of the RrbHLH family.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Hao Y. Zong X. Ren P. Qian Y. Fu A. Basic Helix-Loop-Helix (b HLH) Transcription Factors Regulate a Wide Range of Functions in Arabidopsis Int. J. Mol. Sci.202122715210.3390/ijms 2213715234281206 PMC 8267941 · doi ↗ · pubmed ↗
- 2Toledo-Ortiz G. Huq E. Quail P.H. The Arabidopsis basic/helix-loop-helix transcription factor family Plant Cell 2003151749177010.1105/tpc.01383912897250 PMC 167167 · doi ↗ · pubmed ↗
- 3Atchley W.R. Fitch W.M. A natural classification of the basic helix-loop-helix class of transcription factors Proc. Natl. Acad. Sci. USA 1997945172517610.1073/pnas.94.10.51729144210 PMC 24651 · doi ↗ · pubmed ↗
- 4Atchley W.R. Terhalle W. Dress A. Positional dependence, cliques, and predictive motifs in the b HLH protein domain J. Mol. Evol.19994850151610.1007/PL 0000649410198117 · doi ↗ · pubmed ↗
- 5Jones S. An overview of the basic helix-loop-helix proteins Genome Biol.2004522610.1186/gb-2004-5-6-22615186484 PMC 463060 · doi ↗ · pubmed ↗
- 6Zuo Z.F. Lee H.Y. Kang H.G. Basic Helix-Loop-Helix Transcription Factors: Regulators for Plant Growth Development and Abiotic Stress Responses Int. J. Mol. Sci.202324141910.3390/ijms 2402141936674933 PMC 9867082 · doi ↗ · pubmed ↗
- 7Gao F. Dubos C. The arabidopsis b HLH transcription factor family Trends Plant Sci.20242966868010.1016/j.tplants.2023.11.02238143207 · doi ↗ · pubmed ↗
- 8Feller A. Machemer K. Braun E.L. Grotewold E. Evolutionary and comparative analysis of MYB and b HLH plant transcription factors Plant J. Cell Mol. Biol.2011669411610.1111/j.1365-313X.2010.04459.x 21443626 · doi ↗ · pubmed ↗